
茶葉中虱螨脲等6種農藥殘留量的檢測及結果分析
2022-04-29 00:44:03張聰王秀云陳碩
市場監管與質量技術研究 2022年4期
關鍵詞:固相萃取
張聰 王秀云 陳碩

摘要:為打破發達國家設置關于茶葉中虱螨脲等6種農藥殘留的綠色壁壘,提高我國茶葉出口質量,降低茶葉出口貿易風險,研究建立茶中虱螨脲等6種農藥殘留量的檢測方法。文中首先確定6種農藥的質譜參數,并優化液相條件,然后通過優化實驗條件建立了茶葉中虱螨脲等6種農藥殘留量的液相色譜-串聯質譜檢測方法。方法的標準曲線相關系數、精密度、回收率、檢測限、定量限等指標符合GB/T 27404-2008《實驗室質量控制規范食品理化檢測》要求。用該方法對151批市售樣品進行檢測,除氯噻啉外其他5種農藥均有檢出。該方法便捷、高效、準確、靈敏,可用于茶葉中虱螨脲等6種農藥殘留量的測定。
關鍵詞:茶葉;液相色譜-質譜聯用儀;農藥殘留;固相萃取
Analysis of Detection Results of Six Pesticides Residues Including Lufenuro in Tea
ZHANG Cong WANG Xiu-Yun CHEN Shuo
(1 Fujian Institute for Food and Drug Quality Control, Fuzhou? 35000, Fujian, China)
(2 Fujian Provincial Center for Drug Inspection, Fuzhou 35000, Fujian, China)
Abstract: In order to break the green barriers set up by developed countries about 6 pesticide residues such as lufenuron in tea, improve the quality of China's tea export, and reduce the risk of tea export trade, the detection methods for 6 pesticide residues such as lufenuron in tea were studied and established. Firstly, the mass spectrometry parameters of the six pesticides were determined, and the liquid phase conditions were optimized. Then, a liquid chromatography-tandem mass spectrometry method was established for the determination of six pesticide residues including lufenuron in tea by optimizing the experimental conditions. The correlation coefficient of standard curves, precisions, recoveries, limit of detections, limit of quantifications and other indicators of the method meet the requirements of GB/T 27404-2008 standard: Criterion on Quality Control of Laboratories-Chemical Testing of Food. 151 batches of commercial samples were tested by this method, and all five pesticides except chlorothiline were detected in tea. The method is convenient, efficient, accurate and sensitive, and can be used for the determination of 6 pesticide residues including lufenuron in tea.
Key Words: Tea; Liquid chromatography-tandem mass spectrometry; Pesticide residues; Solid phase extraction
1引言
茶葉是我國傳統特色農產品,在國內外農產品貿易市場中占有十分重要的地位。福建省茶葉產量居全國第一,占全國總產量的20%左右,出口總量位居全國第二[1]。隨著經濟水平的高速發展,人們越來越重視農產品的質量問題。目前,世界各國特別是發達國家都制定了嚴格的茶葉標準,不僅增加了需要檢測的農藥種類,也降低了農藥在茶葉中的最大殘留量,并針對茶葉中農藥殘留量設置了相關的綠色貿易壁壘[2-4],對我國茶葉的出口產生消極影響。據統計,2015年至2020年間,因茶葉農藥殘留對華發出的通告共90次(占總農殘通報比59.5%),僅在2015年一年內,有35批次的茶葉被美國和日本通報為不合格產品,其中因農殘超標的就有29批,占比高達83%[5-6],其中唑蟲酰胺、氯蟲苯甲酰胺、丁醚脲、虱螨脲等農藥被多次通報。因此,我國茶葉出口大國日本和美國及歐盟委員會多次修改茶葉中氯蟲苯甲酰胺、虱螨脲等農藥最大殘留量的限值,修改后的限量值要遠低于之前的數值。由此可見,在茶葉出口貿易中,農藥殘留量是我國目前面臨的最大問題,完善我國現有茶葉中農藥殘留的檢測方法是解決這一問題的途徑之一。
GB 2763-2021《食品中農藥最大殘留限量》作為現行有效的國家標準,其中僅對丁醚脲、唑蟲酰胺和氯噻啉的最大殘留量限值進行規定,丁醚脲和唑蟲酰胺給出了相應的檢驗方法,而虱螨脲、氯蟲苯甲酰胺和甲基硫菌靈仍未給出相應的檢驗方法和限量值。對比我國植物類和茶葉中農殘的其他現行檢驗標準,發現氯蟲苯甲酰胺、虱螨脲、甲基硫菌靈、氯噻啉、唑蟲酰胺在2022年1月1日開始實施的標準GB 23200.121-2021《食品安全國家標準植物源性食品中331種農藥及其代謝物殘留量的測定液相色譜-質譜聯用法》有相應的檢驗方法,丁醚脲在GB 23200.13-2016《食品安全國家標準茶葉中448種農藥及相關化學品殘留量的測定液相色譜-質譜法》有相應的檢驗方法,這6種農藥不能同時檢測,且在標準中氯蟲苯甲酰胺、唑蟲酰胺、虱螨脲、甲基硫菌靈和氯噻啉的定量限均為0.05mg/kg,氯蟲苯甲酰胺、虱螨脲的定量限要高于歐盟規定限量值的0.02mg/kg,氯噻啉的定量限要高于歐盟和日本規定的限量值0.01mg/kg,唑蟲酰胺的定量限要高于歐盟規定的0.01mg/kg[7-10]。由此可見,針對虱螨脲等6種農藥,我國現行的國家標準正在逐步完善,但是也存在著一定的不足,因此建立合適的適用茶葉中虱螨脲等6中農藥殘留量的檢驗方法,對于保障我國茶葉出口及我國人民身體安全具有重要意義。
因此,本研究建立液相-串聯質譜聯用法的同時對殘留進行檢測,對檢測方法的前處理進行調整,并降低了茶葉中虱螨脲等6種農藥殘留檢測的定量限、檢測限。不僅可以對市售產品進行風險監測研究,提高我國茶葉及其制品的食品質量,還可以攻破發達國家設置的關于茶葉中這6種農藥限量值的壁壘,同時為政府監管與企業產品質量改進與提高提供一定依據。
2實驗部分
2.1 試劑與材料
1290 Infinity高效液相色譜儀(美國Agilent公司);AB 5500 Qtrap三重四極桿質譜儀(美國AB公司);CR21N離心機(日本日立公司);MS3型渦旋混合器(德國IKA公司);XSE204型電子天平(0.1mg,瑞士Mettler Toledo公司);HM100刀式研磨儀(北京格瑞德曼公司);實驗用水由Milli-Q超純水器(美國Milipore公司)制得。
甲苯(色譜純,德國默克公司);乙腈(色譜純,美國Fisher公司);甲酸(色譜純,上海阿拉丁公司);石墨化碳/氨基復合固相萃取柱(500mg/500mg,6mL)、弗羅里硅土固相萃取柱(1000mg,12mL)、C18固相萃取小柱(2.0g,12mL)、石墨化碳固相萃取小柱(500mg,6mL)、氨基固相萃取小柱(500mg,3mL)(天津艾杰爾科技公司);氯蟲苯甲酰胺、虱螨脲、甲基硫菌靈、氯噻啉、唑蟲酰胺、丁醚脲標準溶液(濃度均為100μg/mL,天津農業部環境質量監督檢驗測試中心)。
茶葉樣品購自福建各地茶葉市場或超市。
2.2 標準溶液的配制
準確吸取6種標準物質各1mL分別置于6個10mL容量瓶中,用乙腈定容至刻度搖勻,得質量濃度為10μg/mL的標準儲備液,-18℃保存;準確吸取虱螨脲、氯噻啉、丁醚脲標準儲備液各5mL,氯蟲苯甲酰胺、唑蟲酰胺標準儲備液各0.5mL,甲基硫菌靈標準儲備液2.5mL,用乙腈定容至50mL容量瓶中,得混合標準儲備液;根據實驗需求,按照樣品前處理步驟處理空白茶葉樣品制得空白樣品基質,用空白基質溶液配制虱螨脲、氯噻啉、丁醚脲質量濃度為1.0ng/mL、2.0ng/mL、5.0ng/mL、10.0ng/mL、20.0ng/mL、50.0ng/mL,氯蟲苯甲酰胺、唑蟲酰胺的質量濃度為0.1ng/mL、0.2ng/mL、0.5ng/mL、1.0ng/mL、2.0ng/mL、5.0ng/mL,甲基硫菌靈的質量濃度為0.5ng/mL、1.0ng/mL、2.5ng/mL、5.0ng/mL、10.0ng/mL、25.0ng/mL的標準工作溶液。
2.3 樣品前處理
準確稱取10g樣品(精確至0.01g)于50mL離心管中,加入10mL的水浸泡30min,準確加入20mL乙腈溶液,以12000r/min勻漿提取1min,4500r/min離心5min,上清液移入雞心瓶中。再重復提取一次,合并兩次提取液,40℃水浴旋轉濃縮至近干,準確加入5mL乙腈溶解殘余物,待凈化。石墨化碳/氨基固相萃取柱加樣前先用5mL乙腈-甲苯溶液預洗柱,準確移取1mL樣品提取液轉移至固相小柱上,25mL乙腈-甲苯(3:1)溶液洗脫,洗脫液在40℃水浴中旋轉濃縮至近干,加1mL乙腈-水溶液溶解殘渣,經0.22μm微孔濾膜過濾后,待上機測定。
2.4 液相色譜-串聯質譜條件
2.4.1液相條件
ACQUITY UPLC R BEH C18色譜柱(2.1mm×50mm,1.7μm),流動相:0.1%甲酸水(A)-乙腈(B),流速:0.3mL/min,進樣體積:2.0μL,柱溫:30℃。液相色譜梯度洗脫程序:0min~1.0min,70%A;1.0min~5.0min,70%~10%A;5.0min~8.0min,10%A;8.0min~9.0min,10%~70%A;9.0min~11.0min,70%A。柱溫:35℃;流速:0.3mL/min;進樣量:1.0μL。
2.4.2 質譜條件
丁醚脲、甲基硫菌靈、氯噻啉、氯蟲苯甲酰胺、唑蟲酰胺的質譜條件:①離子源:電噴霧電離正負切換模式(ESI±);②掃描模式:多反應監測掃描模式(MRM);③電噴霧電壓:±4500V;④離子源溫度:450℃;⑤氣簾氣壓:20Psi;⑥霧化氣壓:50Psi;⑦輔助氣壓:50Psi;⑧碰撞池入口電壓:9.0V;⑨碰撞室出口電壓:15.0V。
3 結果與討論
3.1 質譜條件優化
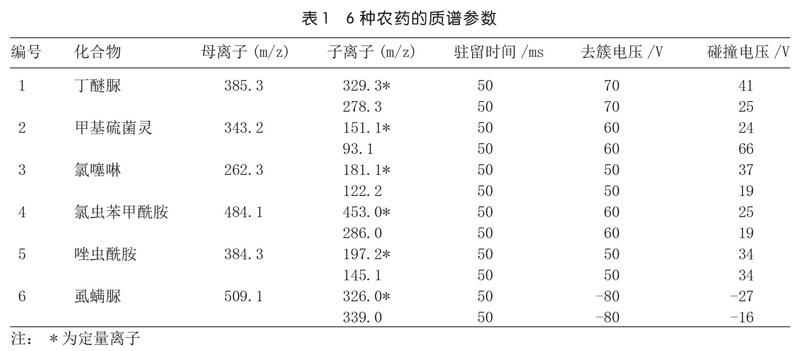
將6種農藥配制成50μg/L濃度的混合標液,在電噴霧正負離子(ESI+/-)模式下進行全掃。結果發現,丁醚脲、甲基硫菌靈、氯噻啉、氯蟲苯甲酰胺、唑蟲酰胺在正離子模式下響應值較高,而虱螨脲在正離子模式下的響應值要比負離子模式下的響應值要低一個數量級,故而前5種化合物在正離子模式下進行檢測,虱螨脲則在負離子模式下進行檢測。采用直接進樣的模式,將50μg/L濃度的混合標液直接注入離子源,在相應的正負離子模式下,先獲得準分子離子峰,結果表明,丁醚脲、甲基硫菌靈、氯噻啉、氯蟲苯甲酰胺、唑蟲酰胺均可形成穩定[M+H]+峰,虱螨脲可形成穩定[M-H]-峰;然后以該分子離子峰為母離子,對準分子離子進行二級質譜分析得相應的離子碎片信息,參照歐盟2002/657/EC的規定[11],每種農藥選擇兩對響應值高且無干擾的特征離子對作為定量和定性的離子對,同時優化了MRM參數,結果詳見表1。
3.2 液相條件優化
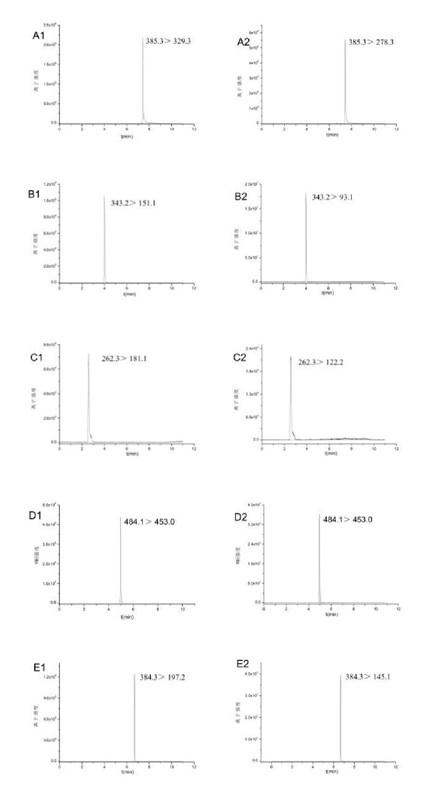
由于6種農藥化合物均含有多個N原子,與色譜柱固定相硅膠上裸露的硅羥基形成氫鍵,造成峰型的拖尾,加入一定量的甲酸,可以抑制化合物與硅羥基之間氫鍵的形成,可以改善峰型,同時甲酸還可提高電噴霧中化合物的離子化效率,因此本試驗選擇在水相中加入一定量的甲酸。反相色譜常用到的有機相為甲醇和乙腈,本試驗比較了以這兩種溶劑為有機相條件下6種化合物的分離度,結果發現以乙腈為流動相的分離效果更好,且在相同條件下乙腈對茶葉中色素、茶多酚等干擾成分的洗脫能力要遠大于甲醇,故本試驗選擇乙腈作為最終的有機相流動相,含有0.1%甲酸水體系為流動相。6種化合物的MRM圖譜詳見圖1。
3.3提取溶劑優化
6種農藥的結構差異較大,但都易溶于乙腈、丙酮、甲醇等有機溶劑。同時茶葉是基質比較復雜的樣品,為了保證提取效率及降低基質中雜質的干擾,選擇常用的乙腈、甲醇、丙酮進行提取效果考察,結果顯示,相對于其他溶劑,乙腈的除雜及提取效果最佳,因其滲透性強,溶解性能良好,不僅能提高6種農藥的回收率,而且能減少提取液中色素、茶多酚等共萃取物的含量,有利于后續的凈化。
3.4凈化條件優化
參照現有的茶葉中農藥檢測標準[12-15],本試驗分別考察了弗羅里硅土固相萃取柱、石墨化碳/氨基復合固相萃取柱、C18-石墨化碳-氨基串聯固相萃取柱對6種化合物回收率的影響。結果發現在經過凈化后,弗羅里硅土固相萃取柱的上機液顏色最深,串聯固相萃取柱的上機液基本無色,石墨化碳/氨基復合固相萃取柱的上機液顏色少帶淺綠,說明串聯固相萃取柱的凈化效果最佳,但是串聯固相萃取柱的上機液回收率低于石墨化碳/氨基復合固相萃取柱,弗羅里硅土柱的顏色深且回收率遠低于其他兩種萃取小柱,結果詳見圖2,故本試驗在SPE凈化法選取石墨化碳/氨基復合固相萃取柱。
3.5 方法學驗證
3.5.1基質效應
在檢測過程中基質效應(ME)是普遍存在的[16],其會影響方法的靈敏度、精密度和準確度,茶葉是一種基質比較復雜的食品,因此需要對所建立的檢測方法中基質效應帶來的影響進行評價。本試驗采用相對響應值的方法[17]來評價6種化合物的基質效應:ME(%)=(1-A/B)×100%,其中A為基質匹配標準溶液的響應值,B為乙腈溶劑標準溶液的響應值。通常認為當ME的絕對值大于20%時,存在較大的基質干擾,當ME為正值是為基質增強,為負值時為基質抑制,由表2可知,6種化合物的ME均為負值,且絕對值均大于20%,說明在液質檢測過程中存在明顯的基質抑制效應,為了提高方法的準確度和靈敏度,本實驗采用空白基質提取液來配置標準溶液,進行外標法定量。
3.5.2 線性、檢出限和定量限
按照2.2配制標準曲線,按照2.4色譜和質譜條件進行檢測,以6種化合物的質量濃度為X軸,定量離子的峰面積為Y軸,繪制工作曲線,得到線性回歸方程及相關系數r,如表2所示,6種化合物的線性關系均良好。以3倍的信噪比(S/N)和10倍的信噪比(S/N)來確定方法的檢出限(limit of detection, LODs)和定量限(limits of quantification, LOQs),如表2所示,丁醚脲、甲基硫菌靈、氯噻啉、氯蟲苯甲酰胺、唑蟲酰胺和虱螨脲的LODs分別為5.0μg/kg、2.5μg/kg、5.0μg/kg、0.5μg/kg、0.5μg/kg、5.0μg/kg,LOQs分別為10.0μg/kg、6.0μg/kg、13.7μg/kg、1.2μg/kg、1.2μg/kg、11.6μg/kg。其中甲基硫菌靈、氯噻啉、氯蟲苯甲酰胺、唑蟲酰胺和虱螨脲的檢出限和定量限要低于現行有效的國家標準,且低于歐盟、美國及日本規定的最大殘留量限值,說明本方法的靈敏度完全滿足實際檢測的要求。
3.5.3 準確度及精密度
本試驗采用加標回收實驗,并以加標回收率來評價方法的準確度,以加標回收的相對標準偏差來評價方法的精密度。先選取不含6種農藥殘留的陰性樣品,分別向其中加入一定量的6種農藥,加標量詳見表3,每個添加水平的實驗平行測定6次,然后計算平均回收率及RSD,結果詳見表3。結果表明,6種農藥的回收率在74.7%~112%之間,RSD在1.8%~8.1%之間,說明本方法具有較好的準確度和精密度,方法的準確性和重復性滿足GB/T 27404-2008《實驗室質量控制規范食品理化檢測》規定的方法回收率偏差、實驗室內變異系數要求,可以用于實際樣品的檢測。
3.6 實際樣品檢測及結果分析
按照最優的實驗條件對151批市場購買的茶葉樣品進行檢測。151批茶葉涵蓋了5大類茶葉(綠茶65批、紅茶20批、白茶12批、黑茶4批、青茶50批),其中有124批次檢出唑蟲酰胺,占比82.1%,20批次檢出氯蟲苯甲酰胺,占比13.2%,27批次檢出丁醚脲,占比17.9%,14批次檢出甲基硫菌靈,占比9.3%,35批次檢出虱螨脲,占比23.2%,氯噻啉未檢出;151批次茶葉中21批次6種農藥殘留均未檢出,占比僅有13.9%,檢出一種農殘的65批次,占比43.0%,檢出兩種農殘的40批次,占比26.5%,檢出三種農殘的20批次,占比13.3%,檢出四種農殘的5批次,占比3.3%。由于歐盟沒有規定茶葉中丁醚脲的最低限量值,按照歐盟最低檢出量默認標準0.01mg/kg來計算,151批茶葉中共20批茶葉丁醚脲不符合歐盟的規定;1批的氯蟲苯甲酰胺和15批的虱螨脲超出歐盟規定的0.02mg/kg;99批次的唑蟲酰胺超出了歐盟規定的0.01mg/kg。
虱螨脲和唑蟲酰胺在5種茶葉中均有檢出。丁醚脲在綠茶、白茶和黑茶中有檢出,其白茶和黑茶的檢出占比相對較大,這可能和茶葉制作工藝有關及丁醚脲易光解、水解有關[18],而綠茶中丁醚脲只能在當年的新茶中檢出,白茶和黑茶不分年份均能檢出,結合相關文獻[18-19],可能是白茶和黑茶在制作工藝中加熱、見光較少,丁醚脲分解較少,所以容易檢出,而針對綠茶、紅茶和青茶制作工藝過程中致使丁醚脲分解而致其未檢出。甲基硫菌靈在綠茶、紅茶和青茶中有檢出,且檢出值均比較小,這和甲基硫菌靈已分解的特性有關,而買到的白茶和黑茶大部分都是陳茶。氯蟲苯甲酰胺除黑茶外均有檢出,但是檢出值相對較低,這跟其本身為新型酰胺類農藥有關,病蟲害的抗藥性較低,故其用藥量較低,且我國還尚未登記其在茶葉種植過程中使用。
結果表明在茶葉種植過程中唑蟲酰胺是普遍使用的一種農藥,且殘留量較大;虱螨脲、丁醚脲、甲基硫菌靈和氯蟲苯甲酰胺存在一定范圍的使用,其中氯蟲苯甲酰胺、甲基硫菌靈、虱螨脲國家標準并未規定限量值,存在一定的食品安全風險。
4 結論
本研究建立了同時檢測茶葉中虱螨脲等6種農藥殘留量的高效液相色譜-串聯質譜測定方法,方法步驟簡便、快速,方法學指標符合GB/T 27404-2008 《實驗室質量控制規范食品理化檢測》的要求。通過本方法對市售的5大類茶葉151批樣品的檢測,結果發現唑蟲酰胺大批量檢出,氯蟲苯甲酰胺、虱螨脲、甲基硫菌靈、丁醚脲少量樣品檢出,只有氯噻啉未發現有檢出,這表明在茶葉種植過程中確有用到上述6種農藥中的5種農藥,唑蟲酰胺是普遍使用的一種農藥,殘留量較大;虱螨脲、丁醚脲、甲基硫菌靈和氯蟲苯甲酰胺存在一定范圍的使用,其中氯蟲苯甲酰胺、甲基硫菌靈、虱螨脲國家標準并未規定限量值,其存在一定的食品安全風險,且會對我國茶葉出口造成一定的影響。本方法可用于茶葉中同時檢測虱螨脲等6種農藥的殘留量,且方法的檢出限和定量低于現行的國家標準和歐盟、美國等發達國家的限量值,可為出口茶葉中這6種農藥檢測提供技術支撐,同時也可為我國現行標準的提高,提供一定的技術支持。另外,可通過方法監測茶葉種植過程中農藥噴施時間和采收時間對茶葉中殘留量的影響,確保在保證茶葉品質和產量的同時,盡可能降低農藥殘留量。
參考文獻
[1]劉新,陳紅平,王國慶.中國茶葉質量安全40年[J].中國茶葉,2019,(12):1-9.
[2]彭威民.綠色壁壘對湖南茶葉出口的影響研究[J].?中國市場,2016,(29):185-186.
[3]周洲.國際市場綠色壁壘下的我國茶葉外貿對策構建[J].?福建茶葉,2017,(7):40-41.
[4]李妍月.歐盟綠色貿易壁壘對中國茶葉出口的影響研究[D].東北財經大學,2016.
[5]張虹艷,楊淑琴,王小喬,高鵬,李赟,邱國玉.2015-2019年歐盟食品和飼料快速預警系統對華食品農獸藥殘留通報的數據分析[J]. 食品安全質量檢測學報,2021,12(7)2600-2606.
[6]朱鳳玲.2013-2017年歐美日韓通報我國不合格茶葉信息匯總與評析[J].中國茶葉,2018,40(6):24-27.
[7]王金鑫.基于歐盟官網通報不合格茶葉信息分析茶葉農殘現狀及應對措施[J].中國茶葉,2018(01):37-39.
[8]丁亦男,童小麟,賴國銀,徐敦明,林立毅,黃旖玨,張志剛.國內外茶葉農藥殘留限量標準與出口茶葉安全研究[J].食品安全質量檢測學報,2019,10(23):8140-8145.
[9]楊梅,羅逢健,周利,樓正云,張新忠,孫荷芝,王新茹,陳宗懋.國內外茶葉農藥最大殘留限量標準比較分析與建議[J].浙江農業學報,2020,?32(1):168-175.
[10]孔紫薇.農藥最大殘留限量標準差異對中國茶葉出口的影響[J]. 福建茶葉,2021(02):36-37.
[11]高海榮.二氧化鋯QuEChERS-高效液相色譜-串聯質譜測定魚肉中2種硝基咪唑及代謝產物[J].食品工業科技,2019,40(4):266-270.
[12]食品安全國家標準茶葉中448種農藥及相關化學品殘留量的測定液相色譜-質譜法:GB 23200.13-2016 [S].
[13]茶葉中農藥多殘留測定氣相色譜/質譜法:GB/T?23376-2009[S].
[14]食品安全國家標準植物源性食品中208種農藥及其代謝物殘留量的測定氣相色譜-質譜聯用法:GB 23200.113-2018[S].
[15]食品安全國家標準植物源性食品中331種農藥及其代謝物殘留量的測定液相色譜-質譜聯用法:GB 23200.121-2021[S].
[16]HE Z,WANG L,PENG Y,et al.Multiresidue analysis?of over 200 pesticides in cereals using?a QuEChERS and gas chromatography-tandem?mass spectrometry-based method[J].Food?chemistry,2015,169:372-380.
[17]孫利東,許秀麗,袁飛,等 .高效液相色譜-串聯質譜法測定牛奶和雞肉中4種激素本底值[J].食品科學,2017,38(22):291-297.
[18]董曉倩,劉松南,劉蕊,黃田田,宗琦,王華,歐陽亞旭. QuEChERS-液相色譜-串聯質譜法測定茶葉中的丁醚脲[J].食品科學,2017,38(08):244-250.
[19]姜楠,劉思潔,崔勇,馬杰.超高效液相色譜-串聯質譜法測定植物源性食品中18種農藥殘留[J].?食品安全質量檢測學報,2021,12(7):2919-2928.
猜你喜歡
分析化學(2016年7期)2016-12-08 00:54:07
中國科技博覽(2016年2期)2016-04-25 14:11:43
湖北工業職業技術學院學報(2016年1期)2016-04-20 17:12:54
分析化學(2015年10期)2015-11-03 07:52:24
食品安全導刊(2015年10期)2015-10-26 04:44:22
安徽農學通報(2015年18期)2015-10-20 00:50:11
安徽農學通報(2015年17期)2015-09-30 00:52:24
分析化學(2015年9期)2015-09-11 07:09:54
肉類研究(2015年5期)2015-08-08 12:46:08
肉類研究(2015年3期)2015-06-16 12:40:36
