丹芪糖脂清顆粒劑提取工藝優(yōu)化*
2022-06-14 02:01:16安紫微彭沙麗李筱玲劉笑洋劉國玉王澳玥姚歡歡
廣州化工 2022年10期
關(guān)鍵詞:實(shí)驗(yàn)
安紫微,彭沙麗,李筱玲,劉笑洋,劉國玉,王澳玥,姚歡歡
(商洛學(xué)院生物醫(yī)藥與食品工程學(xué)院,陜西 商洛 726000)
隨著在社會(huì)生活方式的改變, 代謝綜合征(MS)的發(fā)病率逐年上升, 而且越來越走向年輕化。據(jù)我國相關(guān)調(diào)查表明,我國本病的發(fā)病率已經(jīng)達(dá)到25.9%[1]。代謝綜合征病率呈逐年上升趨勢,且隨年齡升高而增加,是誘發(fā)心腦血管事件、致死、致殘的高危因素[2]。中醫(yī)學(xué)普遍認(rèn)為代謝綜合征的產(chǎn)生及發(fā)展機(jī)制與肝脾有關(guān),認(rèn)為其根本原因?yàn)槠⑻摚瑥亩鴮?dǎo)致水谷不能運(yùn)化所致[3]。因此我們團(tuán)隊(duì)在查閱相關(guān)資料,以《祝氏驗(yàn)方集錦》中的調(diào)節(jié)血脂代謝、降低血糖、調(diào)節(jié)血壓的方劑為基礎(chǔ),經(jīng)過加減組方,擬制成丹芪糖脂清顆粒劑。本方由黃芪、丹參、茯苓、山藥、山楂、決明子、葛根、柴胡、黃芩、玉米須組成,功能健脾除濕,行氣活血化瘀;主治代謝綜合征,高血脂癥、糖尿病、脂肪肝等癥。復(fù)方中主要化學(xué)成分有水溶性物質(zhì)也有脂溶性物質(zhì),傳統(tǒng)的湯劑水煎煮法難以將其脂溶性有效成分提出[4]。本實(shí)驗(yàn)通過醇水雙提法對(duì)丹芪糖脂清顆粒原料藥進(jìn)行提取。以丹參酮ⅡA及丹酚酸B的含量、毛蕊異黃酮葡萄糖苷、葛根素、黃芩苷含量為指標(biāo),以乙醇濃度、料液比、提取時(shí)間為考察因素,采用正交試驗(yàn)法優(yōu)選醇提工藝;以總黃酮含量、總皂苷含量、總多糖含量及干膏率為指標(biāo),以浸泡時(shí)間、加水量、提取時(shí)間為考察因素,采用正交試驗(yàn)法優(yōu)選水提工藝,為丹芪糖脂清顆粒劑中有效成分的提取提供實(shí)驗(yàn)依據(jù)。
1 實(shí) 驗(yàn)
1.1 主要材料
實(shí)驗(yàn)所用的中藥飲片均購買于鶴城大藥房;無水乙醇、葡萄糖,天津市百世化工有限公司;對(duì)照品黃芪甲苷、蘆丁、丹參酮IIA、丹酚酸B、黃芩苷、葛根素、毛蕊異黃酮葡萄糖苷,均購于北京世紀(jì)奧科生物技術(shù)有限公司。
1.2 主要儀器
YLJYE-100數(shù)顯恒溫水浴鍋,邦西儀器科技有限公司;EYEL4OSB-2100旋轉(zhuǎn)蒸發(fā)儀,邦西儀器科技有限公司;LC-10AT高效液相色譜儀,日本島津(中國)有限公司;SI-2002型電子天平,北京丹佛儀器有限公司;DHG-9123A型電熱恒溫鼓風(fēng)干燥箱,上海齊欣科學(xué)儀器有限公司; UV-1780紫外分光光度計(jì), 日本島津(中國)有限公司。
1.3 實(shí)驗(yàn)方法
1.3.1 工藝流程
為了方便實(shí)驗(yàn)操作,按 “丹芪糖脂清顆粒”處方縮小10倍取丹參、黃芪、葛根、黃芩、茯苓、山藥、山楂、決明子、柴胡、玉米須粗粉。先采用乙醇加熱回流法對(duì)丹參、黃芩、黃芪、葛根進(jìn)行回流提取,濾液備用;殘?jiān)c其余藥合并,再采用水加熱回流法對(duì)混合粉末共提,將醇提及水提取液,分別減壓濃縮備用。
1.3.2 乙醇回流提取法條件優(yōu)化
精密稱取同一批飲片粗粉丹參3 g,黃芪3 g,葛根2 g,黃芩1.5 g各9份,浸泡15 min后,90 ℃水浴加熱回流提取,重復(fù)提取2次,合并濾液,減壓濃縮定容至50 mL容量瓶,備用。采用L9(34)正交試驗(yàn)設(shè)計(jì)表進(jìn)行實(shí)驗(yàn),根據(jù)單因素實(shí)驗(yàn)結(jié)果選擇不同乙醇濃度、料液比、提取時(shí)間為參試因子,采用高效液相色譜法測定醇提液中丹參酮ⅡA及丹酚酸B、毛蕊異黃酮葡萄糖苷、葛根素、黃芩苷含量并計(jì)算得率,優(yōu)選項(xiàng)最佳醇提工藝。因素水平表見表1。
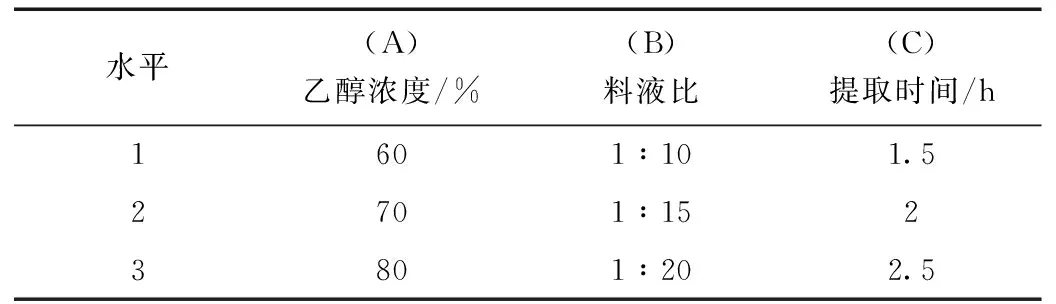
表1 醇提影響因素水平表
1.3.3 水回流提取法條件優(yōu)化
根據(jù)單因素實(shí)驗(yàn)結(jié)果,選擇影響較大的三個(gè)因素浸泡時(shí)間(A)、加水量(B)、提取時(shí)間(C)為考察因素,采用L9(34)正交試驗(yàn)設(shè)計(jì)表安排實(shí)驗(yàn)。具體做法為:按處方量精密稱取藥品粗粉茯苓3 g,山藥3 g,山楂2 g,決明子2 g,柴胡1.5 g,玉米須1.5 g各9份,100 ℃水浴加熱回流提取,重復(fù)提取2次,合并濾液,減壓濃縮定容至50 mL容量瓶,備用。采用紫外分光光度法測定水提液中總黃酮含量、總皂苷含量、總多糖含量,并測定干膏率,優(yōu)選最佳水提工藝。因素水平表見表2。
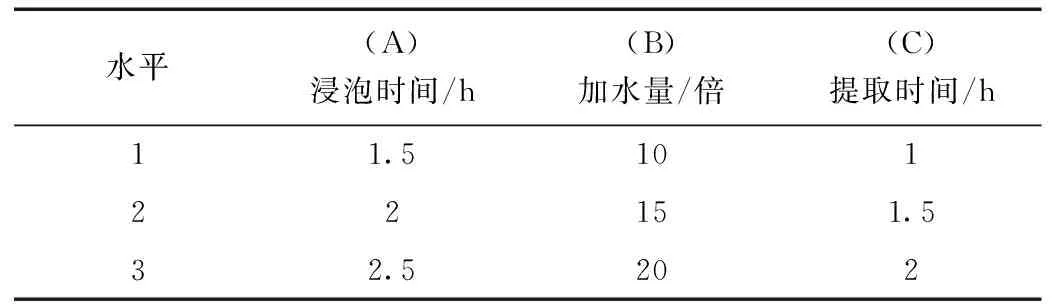
表2 水提影響因素水平表
1.3.4 驗(yàn)證試驗(yàn)
分析正交試驗(yàn)的結(jié)果,得到丹芪糖脂清顆粒顆粒劑提取的最佳工藝條件,按照最佳提取工藝做兩組平行實(shí)驗(yàn)驗(yàn)證試驗(yàn)結(jié)果的準(zhǔn)確性。
1.4 有效成分含量測定
1.4.1 醇提部位有效成分含量測定[5-8]
(1)供試品溶液的制備
取回流提取所得醇提濾液,分別使用0.45 μm微孔濾膜過濾置進(jìn)樣瓶中,即得。
(2)對(duì)照品溶液的制備
丹參酮ⅡA:精密稱取丹參酮ⅡA對(duì)照品適量,精密稱定,置棕色量瓶中,加甲醇制成濃度為37 μg/mL的溶液,即得。
丹酚酸B:精密稱取丹酚酸B對(duì)照品適量,精密稱定,加甲醇-水(8:2)混合溶液制成濃度為176 μg/mL的溶液,即得。
毛蕊異黃酮葡萄糖苷:精密稱取取毛蕊異黃酮葡萄糖苷對(duì)照品適量,精密稱定,加甲醇制成濃度為263 μg/mL的溶液,即得。
葛根素:精密稱 取葛根素對(duì)照品適量,精密稱定,加甲醇制成濃度為655 μg/mL的溶液,即得。
黃芩苷:精密稱取黃芩苷對(duì)照品適量,精密稱定,加甲醇制成濃度為230.4 μg/mL的溶液,即得。
(3)色譜條件
以十八烷基硅烷鍵合硅膠為填充劑;流動(dòng)相:丹參酮ⅡA乙腈-0.02%磷酸,按表3中的規(guī)定進(jìn)行梯度洗脫;丹酚酸B乙腈-0.1%磷酸溶液(22:78);毛蕊異黃酮葡萄糖苷乙腈-0.2%甲酸溶液,按表4中的規(guī)定進(jìn)行梯度洗脫;葛根素0.1%枸椽酸溶液-甲醇,按表5進(jìn)行梯度洗脫;黃芩苷甲醇-水-磷酸(47:53:0.2);柱溫為30 ℃;流速為1 mL/min;檢測波長:丹參酮ⅡA為270 nm;丹酚酸B為286 nm;毛蕊異黃酮葡萄糖苷為260 nm;葛根素為250 nm;黃芩苷為278 nm;進(jìn)樣量:10 μL。

表3 丹參酮梯度洗脫表
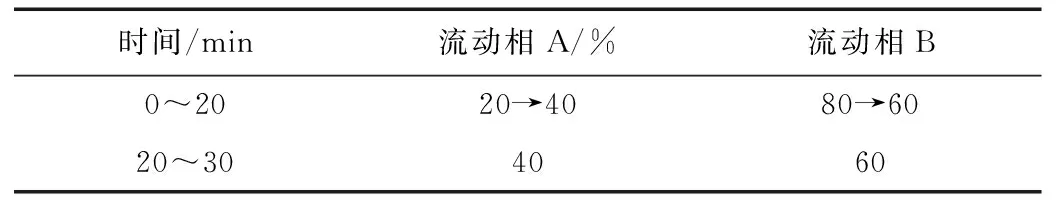
表4 毛蕊異黃酮葡萄糖苷梯度洗脫表

表5 葛根素梯度洗脫表
(4)含量測定:分別精密吸取各對(duì)照品溶液及供試品溶液各10 L,注入液相色譜儀,按照相應(yīng)色譜條件進(jìn)行含量測定。分別計(jì)算樣品中丹參酮ⅡA、丹酚酸B、毛蕊異黃酮葡萄糖苷、葛根素、黃芩苷的含量。
(5)線性關(guān)系考察
分別取丹參酮ⅡA及丹酚酸B、毛蕊異黃酮葡萄糖苷、葛根素、黃芩苷對(duì)照品溶液各2、4、6、8、10 μL,分別注入液相色譜儀,按照各自含量測定方法進(jìn)行檢測。分別以各對(duì)照品溶液濃度(μg/mL)為橫坐標(biāo)(X),峰面積為縱坐標(biāo)(Y),繪制標(biāo)準(zhǔn)曲線,計(jì)算回歸方程。得丹參酮IIA線性回歸方程是y=59208x+2719.9,R2=1;丹酚酸B的線性回歸方程是y=4459.8x-1460.2,R2=0.9999;毛蕊異黃酮葡萄糖苷的線性回歸方程是 y =37463x+27024,R2=0.9978;葛根素的線性回歸方程是 y=40647x-140295,R2=0.9998;黃芩苷的線性回歸方程是 y=25499.1x-4711.1,R2=0.9999;標(biāo)準(zhǔn)曲線如圖1~圖5所示。
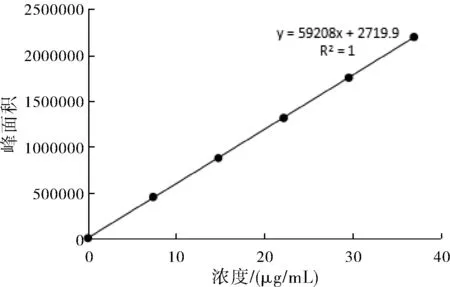
圖1 丹參酮IIA標(biāo)準(zhǔn)曲線
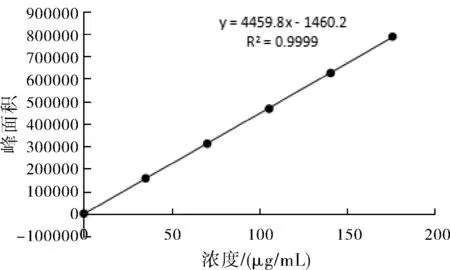
圖2 丹酚酸B標(biāo)準(zhǔn)曲線
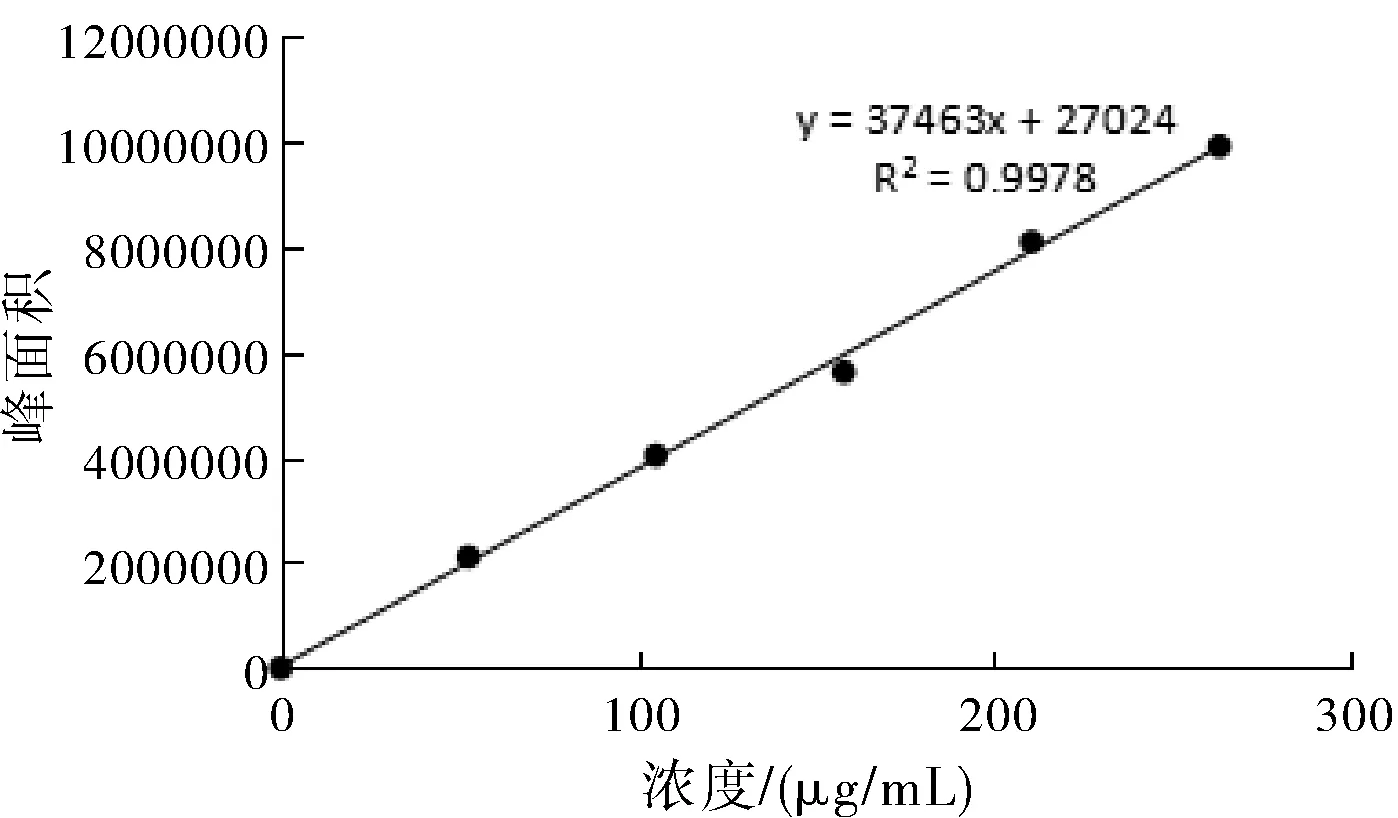
圖3 毛蕊異黃酮葡萄糖苷標(biāo)準(zhǔn)曲線

圖5 黃芩苷標(biāo)準(zhǔn)曲線
(8)綜合評(píng)分
由于實(shí)驗(yàn)的指標(biāo)較多,按照每個(gè)指標(biāo)占比20%,各項(xiàng)指標(biāo)總分為20分。對(duì)各指標(biāo)進(jìn)行綜合評(píng)分,并將各個(gè)指標(biāo)綜合評(píng)分做為這個(gè)試驗(yàn)的總指標(biāo)。以此利用多指標(biāo)綜合評(píng)分法對(duì)實(shí)驗(yàn)結(jié)果作進(jìn)一步的分析,尋找出最優(yōu)的方案或工藝條件。
綜合指標(biāo)=(20/最大丹參酮含量值)×丹參酮含量值+(20/最大丹酚酸B含量值)×丹酚酸B含量值+(20/最大毛蕊花酮苷含量值)×毛蕊花酮苷含量值+(20/最大葛根素含量值)×葛根素含量值+(20/最大黃芩苷含量值)×黃芩苷含量值
1.4.2 水提部位有效成分含量測定[9-11]
(1)供試品溶液制備:取水提濾液做測定供試品溶液。
(2)對(duì)照品溶液的制備
蘆丁:精密稱取蘆丁對(duì)照品20 mg,置100 mL容量瓶中,加95%乙醇50 mL使溶解,然后加50%乙醇稀釋至刻度,即得0.2 mg/mL的對(duì)照品溶液。
黃芪甲苷:精密稱取黃芪甲苷對(duì)照品5.65 mg置25 mL容量瓶中,加無水甲醇使溶解,然后加無水甲醇定容,即得0.226 mg/mL的對(duì)照品溶液。
無水葡萄糖:精密稱取無水葡萄糖對(duì)照品制成濃度為1 mg/mL的對(duì)照品溶液。
(3)標(biāo)準(zhǔn)曲線的制備
總黃酮含量測定以蘆丁為對(duì)照繪制標(biāo)準(zhǔn)曲線;總皂苷含量測定以黃芪甲苷為對(duì)照繪制標(biāo)準(zhǔn)曲線;總多糖含量測定以無水葡萄糖為對(duì)照繪制標(biāo)準(zhǔn)曲線。具體方法如下。
①總黃酮標(biāo)準(zhǔn)曲線的制備:精密量取蘆丁對(duì)照品溶液0、1、2、3、4、5 mL,分別置于10 mL容量瓶中,各加50%乙醇溶液使成5 mL,精密加入5%亞硝酸鈉溶液0.3 mL,搖勻,放置6 min,加入10%硝酸鋁溶液0.3 mL,搖勻,再放置6 min,加入4%氫氧化鈉溶液4 mL,分別用50%乙醇稀釋至刻度,搖勻,放置15 min。以第一瓶作空白,用可見分光光度計(jì)在510 nm處測其吸光度,作標(biāo)準(zhǔn)曲線。總黃酮含量的線性回歸方程是:y=16.041x+0.0457,R2=0.9966,標(biāo)準(zhǔn)曲線線如圖6所示。

圖6 總黃酮含量標(biāo)準(zhǔn)曲線
②總皂苷標(biāo)準(zhǔn)曲線的制備:精密吸取黃芪甲苷標(biāo)準(zhǔn)溶液0、0.15、0.2、0.25、0.3、0.35、0.45 mL于試管中,將溶劑蒸干,加新鮮配制的5%香草醛5 mL(稱取5 g香草醛加冰醋酸100 mL定容至100 mL容量瓶中)和0.8 mL高氯酸,于70 ℃水浴中加熱15 min,冷卻精密加入冰醋酸5 mL搖勻后在30 min內(nèi)用紫外分光光度計(jì),在578 nm波長處測定吸光度。以濃度為橫坐標(biāo),吸光度為縱坐標(biāo)繪制校準(zhǔn)曲線及回歸方程。總皂苷含量的線性回歸方程是y=19.189x-0.0068,R2=0.9937;標(biāo)準(zhǔn)曲線如圖7所示。
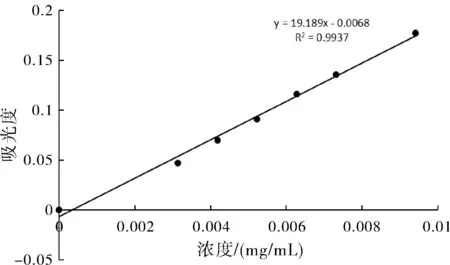
圖7 總皂苷含量標(biāo)準(zhǔn)曲線
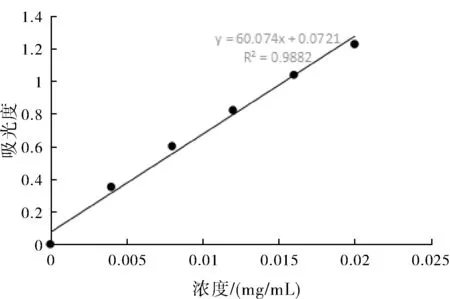
圖8 總多糖含量標(biāo)準(zhǔn)曲線
③總多糖標(biāo)準(zhǔn)曲線的制備:精密量取無水葡萄糖對(duì)照品儲(chǔ)備液 0、1、2、3、4、5 mL 分別置于 50 mL量瓶中用水定容,分別取上述溶液各 2 mL 稀釋液于試管中,迅速加入 1 mL 6% 苯酚,5 mL濃硫酸,靜置 5 min 后置于40 ℃水浴加熱 15 min,再放入冰水中冷卻,靜置至常溫50%乙醇定容至10 mL, 搖勻。在 490 nm 處測定其吸光度,據(jù)濃度與吸光度繪制校準(zhǔn)曲線及回歸方程。總多糖含量的線性回歸方程是y=60.074x+0.0721,R2=0.9882;標(biāo)準(zhǔn)曲線如圖8所示。
(4)含量測定
精密量取水提取液1 mL,3份,分別置于10 mL容量瓶中,分別按照1.4.2(S3)項(xiàng)下①②③標(biāo)準(zhǔn)曲線繪制的方法操作測定吸光度,分別代入標(biāo)準(zhǔn)曲線求濃度,并計(jì)算樣品中總黃酮、總皂苷、總多糖的含量。
(5)干膏率[12]
精密吸取提取液10 mL,分別置于已烘至恒重的蒸發(fā)皿(105 ℃,恒重2 h),蒸發(fā)溶劑至微量水分后,移入恒溫干燥箱恒重3 h后,置干燥器內(nèi)30 min,稱定質(zhì)量并計(jì)算干浸膏得率(兩次稱量結(jié)果誤差不得超過5 mg)。
干膏率=干膏重量×5/凈藥材投入量×100%
(6)綜合評(píng)分
由于實(shí)驗(yàn)的指標(biāo)較多,按照每個(gè)指標(biāo)占比25%,各項(xiàng)指標(biāo)總分為25分,對(duì)各指標(biāo)進(jìn)行綜合評(píng)分,并將各個(gè)指標(biāo)綜合評(píng)分做為這個(gè)試驗(yàn)的總指標(biāo)。以此利用多指標(biāo)的綜合評(píng)分法對(duì)實(shí)驗(yàn)結(jié)果作進(jìn)一步的分析,尋找出最優(yōu)的方案或工藝條件。
綜合指標(biāo)=(25/最大總黃酮含量值)×總黃酮含量值+(25/最大總多糖含量值)×總多糖含量值+(25/最大總皂苷含量值)×總皂苷含量值+(25/最大干膏率值)×干膏率值
2 結(jié)果與討論
2.1 乙醇回流提取工藝優(yōu)化結(jié)果分析
采用L9(34)正交表安排試驗(yàn),分別測定丹參酮ⅡA及丹酚酸B、毛蕊異黃酮葡萄糖苷、葛根素、黃芩苷含量,計(jì)算綜合得分。正交試驗(yàn)結(jié)果見表6,方差分析見表7。
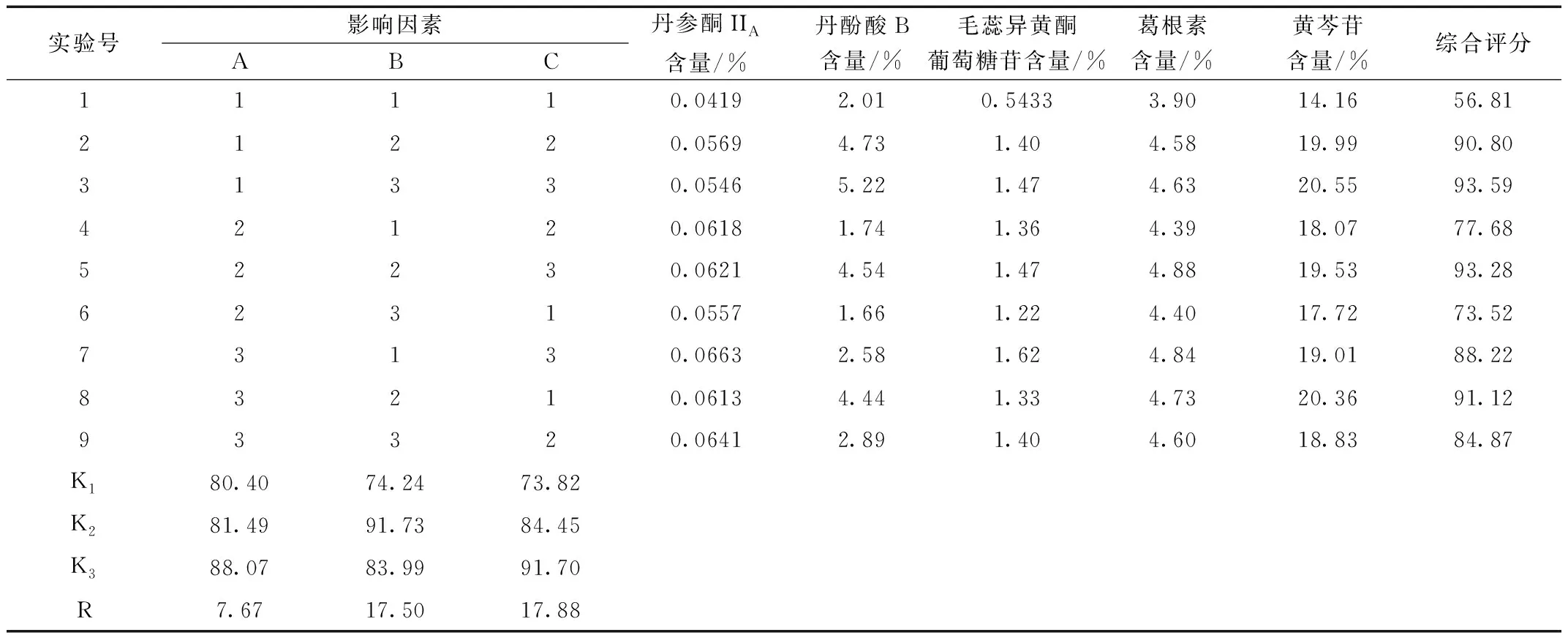
表6 醇提正交試驗(yàn)結(jié)果直觀分析表
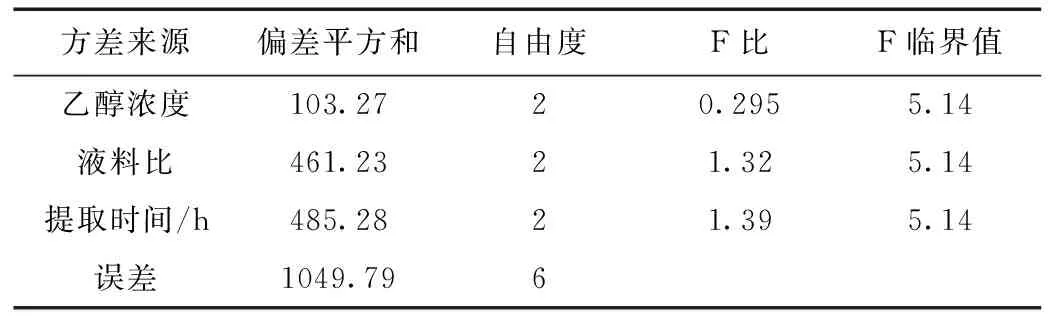
表7 方差分析表
由直觀分析表可知,各因素作用的主次順序依次為提取時(shí)間(C)>料液比(B)>乙醇濃度(A),說明提取時(shí)間對(duì)提取效果的影響最大,其次為料液比及乙醇濃度,由此得出的最佳工藝為 A3B2C3。故最終確定的醇提最佳工藝為A3B2C3,即乙醇濃度80%,料液比為1:15,提取2.5 h。由方差分析表可知,A、B、C三個(gè)因素對(duì)實(shí)驗(yàn)結(jié)果的綜合評(píng)分的影響都不顯著,究其原因可能是本實(shí)驗(yàn)誤差大而自由度小(僅為2),使實(shí)驗(yàn)的靈敏度低,從而掩蓋了考察因素的顯著性。
2.2 水回流提取工藝優(yōu)化結(jié)果分析
采用L9(34)正交表安排試驗(yàn),測定總黃酮含量、總皂苷含量、總多糖含量及干膏率,計(jì)算綜合得分。正交試驗(yàn)結(jié)果見表8,方差分析見表9。
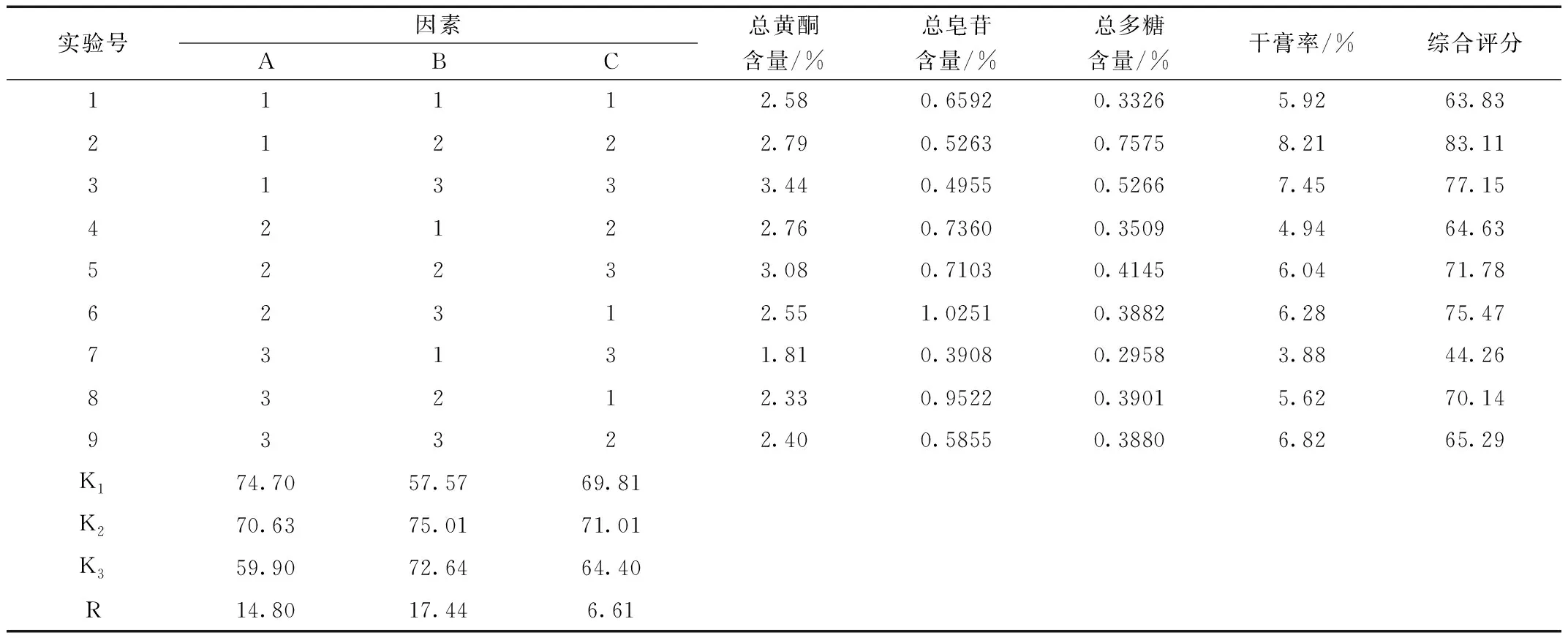
表8 水提正交試驗(yàn)結(jié)果直觀分析表

表9 方差分析表
由直觀分析表可知,各因素作用的主次順序依次為加水量(B)>浸泡時(shí)間(A)>提取時(shí)間(C),說明加水量對(duì)提取效果的影響最大,其次為浸泡時(shí)間及提取時(shí)間,由此得出的最佳工藝為A1B2C2。故最終確定的水提最佳工藝為A1B3C2,即浸泡1.5 h,加20倍量水,提取1.5 h。由表6可知三個(gè)因素對(duì)實(shí)驗(yàn)結(jié)果的綜合評(píng)分的影響都不顯著,究其原因可能是本實(shí)驗(yàn)考察指標(biāo)較多,使實(shí)驗(yàn)的靈敏度低,從而掩蓋了考察因素的顯著性。
2.3 驗(yàn)證試驗(yàn)結(jié)果分析
按處方比例,取丹參、黃芪、茯苓、山藥等十種中藥粗粉,按照1.3.1項(xiàng)下提取工藝流程,分別采用醇提和水體法。醇提法按照加入15倍量的80%乙醇,浸泡15 min,90 ℃提取2.5 h工藝進(jìn)行;水提法按照加20倍量水,浸泡1.5 h,100 ℃提取1.5 h進(jìn)行。醇提液及水提液分別定容至50 mL,備用。重復(fù)實(shí)驗(yàn)2次。個(gè)有效成分測定結(jié)果結(jié)果見表10和表11。

表10 醇提驗(yàn)證實(shí)驗(yàn)結(jié)果表

表11 水提驗(yàn)證實(shí)驗(yàn)結(jié)果表
驗(yàn)證實(shí)驗(yàn)有效成分含量與正交試驗(yàn)結(jié)果最大提取率相差不大,說明結(jié)果穩(wěn)定可靠。
3 討 論
中藥方劑“丹芪糖脂清顆粒劑”處方由10味藥組成,有效成分較為復(fù)雜,在提取過程中不確定因素眾多,如浸泡時(shí)間、提取時(shí)間、液料比、溫度、提取次數(shù)等都各不相同,這些因素會(huì)對(duì)有效成分有一定影響。因此以各種能發(fā)揮藥效的有效成分為指標(biāo),多指標(biāo)綜合測評(píng)實(shí)驗(yàn)結(jié)果,來確定較為科學(xué)可靠的最優(yōu)提取工藝。傳統(tǒng)的水煎煮法提取復(fù)方時(shí),由于藥材較多、成分復(fù)雜,藥材中不易溶于水的成分難以被充分提取,且某些藥物含有揮發(fā)油(如丹參中的丹參酮),水煎煮 提取容易使有效成分揮發(fā)。而本文采取醇水雙提法,能更好的提取脂溶性成分和易揮發(fā)成分。采取回流提取較其他提取方法而言,更加容易操作,對(duì)于工業(yè)化生產(chǎn)也更加經(jīng)濟(jì)。提取所選用的溶劑為乙醇和水,價(jià)格便宜,安全無毒,對(duì)人體無害。
4 結(jié) 論
本文通過在單因素實(shí)驗(yàn)的基礎(chǔ)上進(jìn)行的正交試驗(yàn)優(yōu)選出的丹芪糖脂清顆粒提取工藝,得到最佳提取條件:乙醇回流提取丹參、黃芪、葛根、黃芪時(shí),乙醇濃度80%,料液比為1:15,提取2.5 h時(shí),醇提效果最佳;水回流提取茯苓、山藥、山楂、決明子、柴胡、玉米須時(shí),當(dāng)浸泡時(shí)間為1.5 h,加水量為20倍,提取時(shí)間為1.5 h時(shí),水提效果最佳。
猜你喜歡
作文·小學(xué)低年級(jí)(2025年2期)2025-02-13 00:00:00
小雪花·小學(xué)生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學(xué)低年級(jí)(2024年2期)2024-04-29 00:00:00
作文·小學(xué)低年級(jí)(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(bào)(2022年4期)2022-08-09 08:52:06
中學(xué)生數(shù)理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學(xué))(2019年6期)2019-10-10 01:01:50
發(fā)明與創(chuàng)新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
