以呼吸急促發病的新生兒甲基丙二酸血癥一例并文獻復習
2022-06-17 12:20:40李紫薇劉愛琳顧美群楊佳美楊景暉米弘瑛
云南醫藥 2022年3期
關鍵詞:癥狀
李紫薇,劉愛琳,顧美群,趙 瓊,郭 誠,楊佳美,楊景暉,米弘瑛
(云南省第一人民醫院(昆明理工大學附屬醫院) 兒科,云南 昆明 650032)
甲基丙二酸血癥(methylmalonic aciduria,MMA),是我國有機酸血癥中最常見的類型[1]。因臨床表現缺乏特異性,易出現漏診、誤診。現將1例單純型MMA患兒的臨床診治資料報告如下,希望提高臨床醫師對該病的認識。
1 臨床資料
患兒,男,2日齡,因“呼吸增快”于2021年4月4日由本院產科轉入新生兒科住院治療。系G2P1,38+2周順產娩出,出生體重3.04 kg,出生史無特殊。否認家族性疾病史,否認近親結婚。體格檢查:一般情況及反應欠佳,無特殊面容,口周及肢端稍發紺,呼吸66次/分,伴呻吟、鼻扇,三凹征陽性,雙肺呼吸音粗,未聞及啰音,心、腹及神經系統查體無特殊,CRT<3S。實驗室檢查:WBC 0.65×109/L,Hb 110 g/L,PLT 63×109/L,hsCRP 21.97 mg/L,肝腎功能基本正常,血氨791 umol/L;動脈血氣分析pH值7.17,堿剩余-21.92 mmol/L,HCO3-3.6 mmol/L,Lac 15.87 mmol/L;同型半胱氨酸正常,血糖正常,血培養陰性。
診治經過及結局:入院后予輔助通氣、抗感染、配方奶喂養、補液、糾酸等治療;生后第5 d出現喂養不耐受伴腹脹,予禁食、補液;生后第6 d出現抽搐,復查血常規提示全血細胞減少。因患兒存在呼吸困難、嚴重酸中毒、喂養不耐受、抽搐、全血細胞減少等癥狀,懷疑代謝性疾病可能,取得患兒父母同意后,送檢血/尿有機酸及基因檢查;生后第9 d出現肺出血,予高頻呼吸機輔助通氣,同時予止血、補充凝血因子、改善循環等綜合治療,但患兒病情仍進行性加重,搶救無效死亡。
補充結果:血串聯質譜:丙酰肉堿、丙酰肉堿/乙酰肉堿明顯升高;尿有機酸分析:甲基丙二酸、甲基枸櫞酸、3-羥基丙酸明顯升高,均提示甲基丙二酸血癥。基因檢查:患兒存在MUT基因c.1777G>T(p.E593*)雜合突變和c.1159A>C(p.T387P)雜合突變,見圖1。患兒父母均無臨床表型,符合常染色體隱性遺傳。
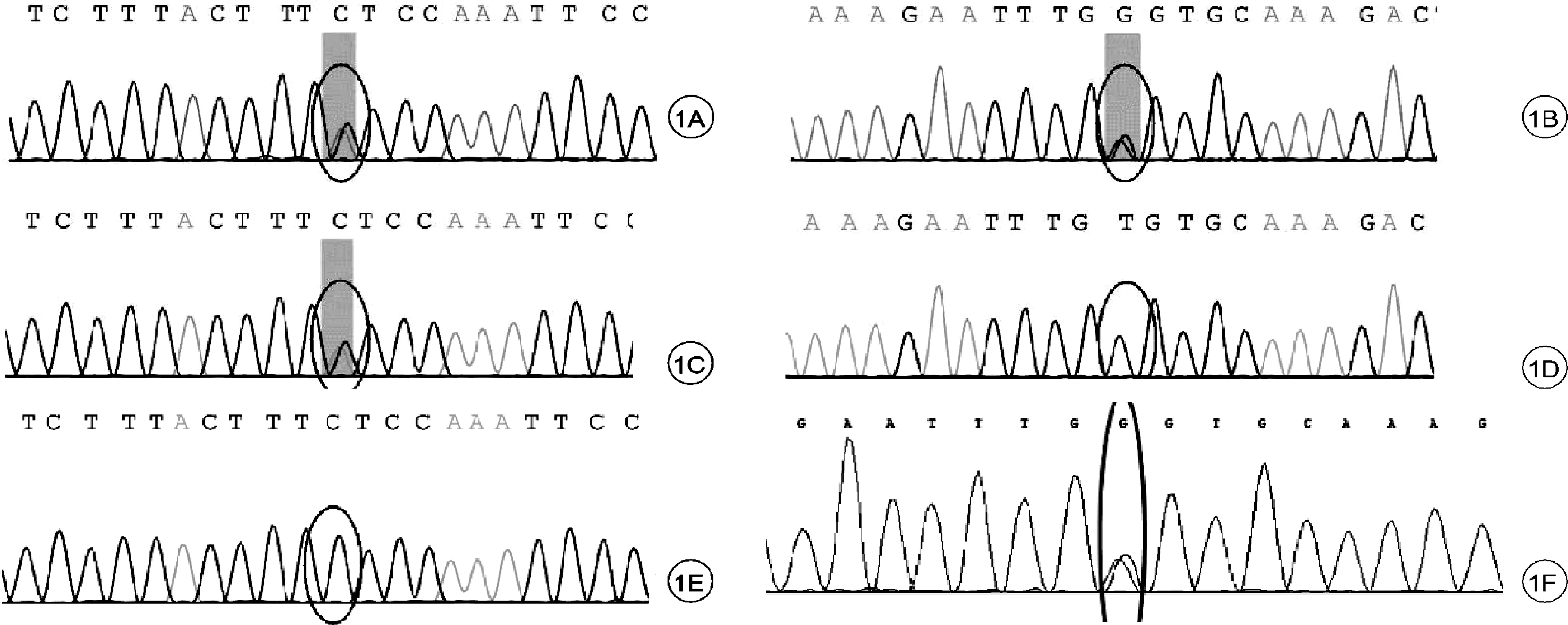
圖1 MUT基因突變Sanger測序驗證圖
2 討論
MMA是由于機體甲基丙二酰輔酶 A 變位酶(methylmalonyl-CoA mutase,MCM)存在缺陷或其輔酶腺苷鈷胺素代謝障礙,導致甲基丙二酰輔酶 A 轉換為琥珀酰輔酶 A的過程發生障礙,致使機體內甲基丙二酸及其相關代謝產物在體內大量堆積,造成神經、心臟、腎臟及免疫等多個系統損傷。根據是否合并同型半胱氨酸增高可分為單純型及合并型。其中由于MCM缺陷而引起的為單純型MMA,占總體的30%[2]。本例患兒基因檢查結果提示為MUT0型,即完全性MCM缺陷型,生化結果顯示同型半胱氨酸正常,符合單純型MMA。
MMA在新生兒期發病者可能在生后 2~3 d內出現癥狀,表現為精神不佳、喂養困難、呼吸急促、嘔吐、抽搐、甚至意識障礙、昏迷等。本例患兒生后第2 d發病,以呼吸急促為首要癥狀,病程中出現精神不佳、喂養困難、抽搐癥狀,均符合上述表現。
MMA多為單基因引起的常染色體隱形遺傳病,至今為止已發現MMACHC、MUT等7種致病基因。一項多中心研究發現:在單純型MMA中,MUT基因變異是主要病因(90.3%)[1,2]。本例患兒為單純型MMA,基因結果為MUT基因復合雜合突變,與既往研究相一致。
人體內的MCM由MUT基因編碼,MUT基因定位于染色體6p21,由13個外顯子組成,分布在35kb范圍內,目前發現的MUT基因變異有200余種,多為單個堿基替換(約84%)[3]。
本例患兒基因結果提示存在MUT基因c.1777G>T(p.E593*)雜合突變和c.1159A>C(p.T387P)雜合突變,兩個位點均為已知的致病變異。其中c.1777G>T(p.E593*)為無義突變,在Yue Yu等人的報道中發現攜帶此突變的患兒對維生素B12治療無反應[4]。c.1159A>C(p.T387P)報道較多,其中在一項針對MMA患兒及基因變異分析中,c.1159A>C為最常見的變異位點[5]。
綜上所述,本例患兒出現呼吸急促、精神不佳、喂養不耐受、抽搐、全血細胞減少等癥狀,但各癥狀均無特異性,因此在疾病早期容易出現漏診、誤診。通過對本病例分析及相關文獻復習,提醒我們在臨床上對存在多器官、多系統受累者應早期警惕代謝性疾病,盡早完善血液及尿液氨基酸檢測,盡可能完善相關基因檢查,爭取做到早發現、早診斷、早治療,提高救治成功率。
利益沖突聲明:所有作者均聲明本研究不存在利益沖突。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
