優化近乎PAM-less腺嘌呤堿基編輯器提升編輯效率
2022-06-27 02:03:02周小鈺王曉月
基礎醫學與臨床 2022年6期
關鍵詞:效率
周小鈺,王曉月
(中國醫學科學院基礎醫學研究所 北京協和醫學院基礎學院 生物化學與分子生物學系,北京 100005)
在已知的人類致病遺傳變異中,約50%是由單個核苷酸突變引起的[1],因此,開發高效的點突變糾正工具是人類遺傳疾病研究和治療的重要目標。目前,單堿基編輯技術將Cas9蛋白與脫氨酶結合,可以在sgRNA的引導下對基因組靶位點進行單核苷酸突變[2],其中腺嘌呤堿基編輯器(ABE)能將靶位點的腺嘌呤(A)轉變為鳥嘌呤(G)[3]。該技術不會引起DNA雙鏈斷裂[2],具有更高的安全性。
然而單堿基編輯技術也存在一定的局限性:首先,最常用的SpCas9蛋白識別靶位點時必須首先識別NGG PAM序列[4],極大的限制了其應用范圍。其次,編輯效率較低也是阻礙該技術在基因治療領域發揮作用的重要因素[5]。本研究所采用的PAM-less ABE[6]在SpCas9蛋白上進行了突變使其幾乎不受PAM序列的限制,擴大了在基因組的編輯范圍。在此基礎上通過對ABE其他元件如脫氨酶和連接肽進行替換改造,在基因組上非NGG PAM的位點進行編輯效率驗證實驗,從中初步評估ABE的編輯效率。
1 材料與方法
1.1 材料
HEK 293T人胚腎細胞系(國家生物醫學實驗細胞資源庫);DMEM高糖培養基(Hyclone公司);gRNA序列(擎科生物科技公司);NeofectTM轉染試劑(零客創智公司);lentiGuide-Puro 載體、pCMV-T7-ABEmax(7.10)-SpRY-P2A-EGFP載體(簡稱為SpRY-ABEmax)(Addgene公司);胎牛血清(Gibco公司);Opti-MEM(Life公司);Tryptone、Yeast Extract(OXOIO公司);Agar(鼎國昌盛公司);Trans5α 化學感受態細胞(全式金生物技術公司); FastAP、MluⅠ、BglⅡ、BsmBⅠ(Thermo Scientific公司);T4 DNA Ligase、T4 PNK、Gibson Assembly? Master Mix、NEBNext? UltraTMⅡ Q5? Master Mix(NEB公司); 瓊脂糖凝膠DNA回收試劑盒、DNA maker D2000、質粒提取試劑盒、基因組DNA提取試劑盒(上海天根生化科技公司)。
1.2 方法
1.2.1 PAM-less ABE的改造:首先利用MluⅠ和BglⅡ雙酶切SpRY-ABEmax載體,膠回收大片段為載體骨架,然后通過克隆突變得到Tad-8e,退火合成PAPAPA連接肽。利用重疊延伸 PCR將各片段連接,通過Gibson 組裝將片段和載體骨架連接成環并轉化至大腸桿菌感受態細胞,挑單克隆進行Sanger 測序驗證組裝是否正確,提取成功組裝的質粒并轉染HEK 293T細胞, 48 h后觀察綠色熒光蛋白表達情況以檢驗ABE在細胞中是否表達。
1.2.2 sgRNA 表達載體克隆構建:將lentiGuide-Puro 載體的scaffold部分改造以提升sgRNA轉錄效率,具體方法見參考文獻[7]。使用BsmBⅠ酶切lentiGuide-Puro,保留大片段作為載體骨架,將sgRNA的兩條oligo退火和磷酸化,使用T4連接酶lentiGuide-Puro載體骨架與sgRNA片段連接,轉化至感受態細胞并挑單克隆進行Sanger測序,提取成功連接sgRNA的質粒(表1)。

表1 不同基因組位點的sgRNA序列Table 1 sgRNA sequences at different genomic sites
1.2.3 ABE和sgRNA表達質粒共轉染HEK 293T細胞:取60萬HEK 293T細胞鋪于6孔板,第2天觀察當細胞匯合至90%時進行下一步實驗。在1.5 mL EP管內添加 200 μL Opti-MEM培養基,每管加入2.5 μg ABE表達質粒和1.5 μg sgRNA表達質粒并加入4 μL NeofectTM轉染試劑,混勻室溫放置30 min后加入6孔板。轉染24 h后加入1.5μg/mL的嘌呤霉素并于72 h后流式分選表達綠色熒光蛋白的細胞,將其凍存于負80或立即進行實驗。
1.2.4 提取細胞基因組并使用PCR擴增技術在預期產生編輯的位點進行擴增和Sanger測序:使用BEAT工具[8]對Sanger測序的.ab1格式文件進行分析預估各版本ABE在同一編輯位點的編輯效率,每組設置兩次或3次重復實驗(表2)。

表2 PCR所用引物的序列Table 2 Sequence of primers used in PCR
1.3 統計學分析
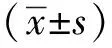
2 結果
2.1 PAM-less ABE轉染HEK 293T細胞的編輯效果
在基因組上PAM序列為GCTG的位點進行A到G的編輯,Sanger測序結果顯示在第3位和第5位(前間隔序列protospacer的第1位至第20位為sgRNA與基因組DNA互補序列)的編輯效率分別為32.5%、70.5%(圖1)。
2.2 PAM-less ABE的改造及驗證
以SpRY-ABEmax為初始版本,在此基礎上替換Tad-8e脫氨酶和PAPAPA連接肽分別構建SpRY-ABE8e和SpRY-ABE8e-PAPAPA(圖2A),將各版本PAM-less ABE分別轉染HEK 293T細胞,48 h后熒光顯微鏡下觀察,帶有綠色熒光的細胞即為表達PAM-less ABE的細胞(圖2B)。
2.3 改造各版本PAM-less ABE編輯效率的比較
與對照組SpRY-ABEmax相比:1)SpRY-ABE8e在PAM序列為GACT和CAAC位點的編輯效率提升,編輯窗口也擴大;2)SpRY-ABE8e-PAPAPA提升了這兩個基因組位點的中心位置(GACT-6A或CAAC-5A)的編輯效率,同時編輯窗口內的旁編輯情況相較于SpRY-ABE8e降低(圖3);3)SpRY-ABE8e-PAPAPA在基因組PAM序列為GACG的兩個位點的整體編輯效率更高(圖4)。
2.4 PAM-less ABE對Clinvar致病突變位點的編輯效果
SpRY-ABEmax在Clinvar數據庫中BRCA1:p.L1705P和BRCA2:p.D189G兩個位點的編輯效率分別為53%和26%,SpRY-ABE8e-PAPAPA在對應兩個位點的編輯效率分別為60%和35%(圖5)。

protospacer (black and red) and PAM (blue) sequences are located above the histogram; target As are indicated in redwithin the protospacer

圖2 各版本PAM-less ABE的構建 (A)和轉染HEK 293T細胞的綠色熒光蛋白表達驗證(B)

protospacer (black and red) and PAM (blue) sequences are located above the histogram; target As are indicated in red within the protospacer;*P<0.05 compared with SpRY-ABEmax

protospacer (black and red) and PAM (blue) sequences are located above the histogram; target As are indicated in red within the protospacer;*P<0.05 compared with SpRY-ABEmax

*P<0.05 compared with SpRY-ABEmax圖5 PAM-less ABEs 在BRCA1和BRCA2基因上構造致病突變的編輯效率Fig 5 Base editing efficiency of PAM-less ABEs to construct pathogenic mutations in BRCA1
3 討論
基于CRISPR-Cas系統的單堿基編輯技術可以直接在細胞基因組內實現單個堿基的轉換類突變,這種方式避免了DNA雙鍵斷裂可能對細胞基因組造成的損傷[8]。但由于PAM序列識別位點的限制,目前主流的堿基編輯器受到NGG PAM序列的限制,只有當靶位點下游13~17 bp范圍內存在NGG PAM序列時才能進行編輯,限制了應用范圍[4]。本研究中的SpCas9變體SpRY幾乎不受PAM序列限制,擴大了基因組的編輯范圍[6]。然而ABEmax編輯效率較低[9],因此需要對其進行改造來提升編輯效率以實現更多的應用。
本研究首先驗證了SpRY-ABEmax能對非NGG PAM的基因組位點進行編輯,然后嘗試替換活性較高的腺嘌呤脫氨酶來改造新版本的PAM-less ABE。根據已有研究顯示,最新的ABE8e 進行脫氨的速率快了大約1 100倍[10],在本研究中將ABE8e與SpRY進行組合構建SpRY-ABE8e并與sgRNA表達質粒共轉染HEK 293T細胞,通過對幾個非NGG PAM的基因組位點進行編輯效率驗證實驗發現:相較于SpRY-ABEmax,SpRY-ABE8e提升了對應基因組位點的編輯效率,但ABE-8e的高活性導致了編輯窗口擴大,從而產生更多的旁編輯,考慮到這個問題,根據之前的一項研究,縮短連接肽會在一定程度上限制編輯窗口[11],因此本研究采用了PAPAPA連接肽構建SpRY-ABE8e-PAPAPA,在一定程度上保持了中心位點編輯效率的同時限制了ABE8e的編輯范圍,進一步將該版本ABE應用在ClinVar致病位點的編輯上[12],結果顯示相較于對照組,SpRY-ABE8e-PAPAPA提升了對應位點的編輯效率。本研究所改造的PAM-less ABE有助于進一步對人類單核苷酸變異位點進行研究,由于突破了PAM序列的限制,可以在更多基因組位點進行疾病模型的構建或疾病治療模型的構建,在基因治療領域有廣闊的前景。
綜上所述,本研究證實通過替換活性更高的脫氨酶Tad8e可提升編輯效率;進一步可縮短脫氨酶與SpRY蛋白之間的連接肽來限制編輯窗口。該研究為今后基礎研究和基因治療應用提供了更多選擇的堿基編輯工具。
猜你喜歡
瘋狂英語·初中天地(2021年5期)2021-07-21 02:24:28
甘肅教育(2020年14期)2020-09-11 07:57:42
中學生數理化(高中版.高考數學)(2020年5期)2020-06-02 09:19:08
商周刊(2017年9期)2017-08-22 02:57:49
遼寧經濟(2017年6期)2017-07-12 09:27:16
中國衛生(2016年9期)2016-11-12 13:27:54
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國洗滌用品工業(2015年7期)2015-02-28 19:02:38
電子設計工程(2015年12期)2015-02-27 12:06:10
中國衛生(2014年11期)2014-11-12 13:11:32
