STARD3在人類表皮生長因子受體2陽性乳腺癌中的研究進展
2022-06-30 03:17:36鄭彩華林丹霞
汕頭大學醫學院學報 2022年2期
關鍵詞:乳腺癌
鄭彩華,林丹霞
(汕頭大學醫學院附屬腫瘤醫院,廣東 汕頭 515041)
乳腺癌是女性最常見的惡性腫瘤,目前已超越肺癌成為女性癌癥相關死亡的首要原因[1]。15%~20%的乳腺癌存在人類表皮生長因子受體2(human epidermal growth factor receptor 2,HER2)基因擴增或過表達,稱為HER2陽性乳腺癌[2]。HER2陽性乳腺癌易發生耐藥和轉移。目前有觀點認為HER2基因擴增可能是促進HER2陽性乳腺癌細胞轉移和耐藥的重要原因之一,靶向抑制HER2基因可增強HER2陽性乳腺癌治療療效[3]。目前,類固醇合成急性調節相關脂質轉運蛋白3(steroidogenic acute regulatory protein related lipid transfer domain containing 3, STARD3) 與HER2基因同源且與其共擴增、共過表達,且STARD3在侵襲性乳腺癌中的轉錄水平和蛋白水平均有升高,與乳腺腫瘤局部復發、轉移、患者總生存期顯著相關[4-6]。這提示STARD3過表達可能與HER2陽性乳腺癌預后不良相關。本文就STARD3調節細胞內膽固醇分配過程以及與HER2陽性乳腺癌的關系進行綜述,根據現有的實驗,重點分析STARD3參與HER2陽性乳腺癌轉移的潛在機制。
1 STARD3概述
STARD3又稱轉移性淋巴結蛋白64,最早發現于在浸潤性乳腺癌中高表達,其基因定位在17 q11-q21區域上,與HER2基因共擴增。STARD3蛋白有兩個不同結構域:N端MENTAL結構域和C端START結構域(圖1A)。MENTAL結構域可將該蛋白靶向定位于晚期內體膜上[7]。內體是由細胞膜內陷形成,通過酸化和物質交換發展為晚期內體[8]。START結構域含有一個疏水的膽固醇結合口袋,以1∶1的比例結合膽固醇[9]。在兩末端之間存在一個中心基序(酸性管道中的兩種苯丙氨酸),該基序通過磷酸化與內質網上的囊泡相關膜蛋白相關蛋白相互作用,在內質網和晚期內體之間創建緊密的附著區域[10](圖1B)。
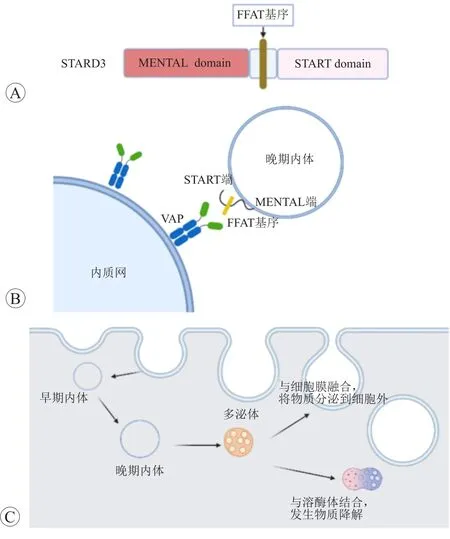
圖1 STARD3的結構和功能
2 STARD3參與細胞內膽固醇分配
膽固醇攝入細胞后通過囊泡轉運和非囊泡轉運兩種方式進行分配[11]。STARD3參與細胞內膽固醇的非囊泡轉運過程,通過MENTAL結構域將其靶向定位于細胞內的晚期內體膜;并通過C端的疏水性膽固醇結合口袋結合細胞內游離膽固醇[7,9]。隨著晚期內體向溶酶體或細胞膜的轉移,將膽固醇轉運至溶酶體或者細胞膜上(圖1C),參與細胞內膽固醇的分配過程,但不參與細胞內膽固醇的穩態平衡調節[8]。
3 STARD3與HER2陽性乳腺癌的關系
研究表明存在HER2擴增或過表達的乳腺癌患者,HER2擴增的同時,17q12-q21上存在多個基因片段共擴增和共過表達[12]。有觀點認為擴增子可能是促進HER2陽性乳腺癌細胞轉移和耐藥的重要原因之一,靶向抑制HER2時,如果同時抑制擴增子可增強抗HER2治療的療效[3]。RNA印跡法分析顯示在93例浸潤性原發性乳腺癌中有14例和在7例乳腺癌轉移灶中有3例表達了2.1 kb的STARD3轉錄本[13]。Cai等[5]通過收集146份乳腺癌標本進行免疫組化檢測,發現與正常乳腺標本相比,STARD3在侵襲性乳腺癌中的轉錄水平和蛋白水平均有升高,且與患者的腫瘤局部復發、轉移、總生存期顯著相關。Vassilev等[4]收集芬蘭1991—1992年確診乳腺癌的病理標本進行免疫組化檢測,發現大約10.2%(92/895)的乳腺癌存在STARD3過表達,患者總生存期較差。然而,以上研究都沒有根據乳腺癌分子分型進行更細致的研究。
Fararjeh等[6]通過生物信息學網站進行大數據挖掘發現在HER2陽性乳腺癌患者中,STARD3表達水平較高,且STARD3高表達預示著總生存期、無復發生存期和無疾病轉移生存期更差。這提示STARD3過表達可能與HER2陽性乳腺癌預后不良相關,然而,現有的研究尚未能闡述STARD3是如何影響HER2陽性乳腺癌轉移的,本文就現有的研究進行整理和總結,提出幾種可能機制。
4 STARD3參與HER2陽性乳腺癌轉移的可能機制
4.1 通過影響內質網膽固醇含量從而促進乳腺癌轉移
Vassilev等[4]對乳腺癌患者病理標本的免疫組化研究發現STARD3過表達時,3-羥基-3-甲基戊二酰輔酶A還原 酶 (3-hydroxy-3-methylglutaryl-coenzyme A reductase,HMGCR)蛋白表達也升高;細胞學水平研究發現STARD3表達增加,HMGCR表達也增加,同時細胞內質網上膽固醇含量下降。Wilhelm等[14]進一步研究發現STARD3和內質網上囊泡相關膜蛋白相關蛋白的相互作用能促進內質網上膽固醇向晚期內體膜轉移,目前已證實這種甾醇轉移是通過苯丙氨酸磷酸化進行的。而現有研究證實內質網膽固醇含量下降可促進內質網固醇調節元件結合蛋白2進入細胞核并啟動包括HMGCR在內的甾醇生物合成蛋白質的表達[15]。而HMGCR表達增加與乳腺癌的預后不良相關[16]。這提示STARD3可能通過調節內質網上膽固醇含量來影響固醇調節元件結合蛋白2的表達,從而影響乳腺腫瘤的轉移。然而目前未見相關報道證明抑制STARD3可直接減少HMGCR的轉錄本數量。
4.2 通過引起線粒體膽固醇蓄積從而促進乳腺癌轉移
Charman等[17]在晚期內體蛋白NPC1缺陷細胞中研究發現STARD3調節膽固醇向線粒體的運輸并可引起膽固醇在線粒體外膜蓄積。Balboa等[18]進一步發現STARD3過表達時,細胞線粒體中活性氧(reactive oxygen species,ROS)的產生增加、線粒體出現碎片化情況;下調STARD3表達可改善線粒體功能。Zhou等[19]在3T1-L1細胞中研究STARD3的表達與ROS的關系,敲除STARD3基因時發現細胞線粒體ROS水平降低。
上述的研究表明STARD3表達與線粒體膽固醇和ROS水平有關。而過量ROS的產生可通過氧化還原信號促進腫瘤細胞的增殖、存活和侵襲[20]。這提示STARD3可能通過促進膽固醇向線粒體轉運,引起線粒體上膽固醇蓄積導致細胞內ROS增加,進而促進腫瘤細胞的增殖、轉移。然而,目前關于STARD3如何促進膽固醇轉移至線粒體上的機制尚未明確。
4.3 通過激活FAK-Src信號通路從而促進乳腺癌轉移
Cai等[5]通過構建人乳腺癌STARD3基因敲除細胞株進行研究,發現STARD3基因敲除細胞的黏附能力明顯增強。同時對2種基質黏附調節分子樁蛋白和黏著斑激酶(focal adhesion kinase,FAK)進行染色,與野生型細胞和轉染對照組細胞相比,發現STARD3基因敲除細胞株中FAK染色顯著增強,但是樁蛋白的染色沒有差異。蛋白質印跡法分析顯示,STARD3基因敲除細胞株中FAK蛋白的水平增加,這表明STARD3可能通過FAK參與細胞基質黏附調節。同時,FAK抑制劑實驗也進一步支持這一觀點。
Vassilev等[4]在完全培養基中對STARD3過表達的細胞進行培養,發現與對照組相比,STARD3細胞中FAK的磷酸化水平雖無顯著差異,但撤去生長刺激后,細胞中FAK的含量和磷酸化水平都顯著增加。這個結果反向驗證了Cai等[5]的研究。Src是細胞黏附的關鍵調控因子,進一步檢測STARD3過表達細胞中Src含量,發現Src水平明顯升高。通過抗Src染色分析發現17.2%的乳腺癌腫瘤樣本磷酸化Src染色為陽性,并且Src磷酸化與STARD3蛋白表達相關。
FAK-Src信號通路與癌細胞的遷移密切相關,由此推測STARD3過表達可能通過激活FAK-Src信號通路從而促進腫瘤轉移。
5 總結與展望
腫瘤的預后不良與其易轉移和耐藥密不可分。在HER2陽性乳腺癌中,HER2表達增加的同時STARD3轉錄本、蛋白質表達也在增加。我們推測過表達的STARD3蛋白可通過降低或增加細胞內質網/線粒體生物膜上膽固醇含量或者通過減少FAK的生成導致細胞生長失控或細胞黏附性下降從而引起腫瘤的進展,且研究發現抑制STARD3可有效地抑制腫瘤細胞的增殖生長。這提示STARD3可能是一個預測乳腺癌轉移的風險指標及潛在的靶向治療靶點。然而目前臨床上尚未研究出相關的靶向藥物,這需要我們進行更多的探索和研究。
猜你喜歡
中老年保健(2022年6期)2022-08-19 01:41:48
現代臨床醫學(2022年1期)2022-02-12 02:04:58
甘肅科技(2020年20期)2020-04-13 00:30:42
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
中國生殖健康(2019年5期)2019-01-06 09:16:40
幸福家庭(2019年14期)2019-01-06 09:15:38
祝您健康(2018年5期)2018-05-16 17:10:16
癌癥進展(2016年9期)2016-08-22 11:33:20
中國組織化學與細胞化學雜志(2016年4期)2016-02-27 11:16:08
