高效液相色譜法測定血漿中替考拉寧的血藥濃度
2022-08-25 08:48:22王欽姚燕
中國當代醫藥 2022年20期
王 欽 姚 燕
1.南通大學附屬醫院藥學部,江蘇南通 226001;2.上藥控股南通有限公司,江蘇南通 226001
替考拉寧臨床可用于耐甲氧西林金黃色葡萄球菌和腸球菌感染的治療,適應證主要是膿毒癥、膿毒性關節炎、嗜中性粒細胞減少、膿毒性心內膜炎、骨髓炎急性發作等,且已知的不良反應發生率低。替考拉寧口服無效,與萬古霉素比較,其半衰期明顯延長至80~168 h,在體內約97%以原型經腎排泄,其余經肝臟代謝。現有的替考拉寧血藥濃度檢測方法主要有:高效液相色譜法(high performance liquid chromatography,HPLC)、液相色譜-串聯質譜聯用法、微生物檢定法、熒光偏振免疫法、膠束電動毛細管色譜法等,其中,HPLC 法具有操作簡單、靈敏度高、專屬性好、成本較低廉等優勢。治療非嚴重細菌感染替考拉寧的穩態血藥谷濃度需達到10~25 μg/ml,方可達到治療效果,而治療嚴重細菌感染則需達到15~30 μg/ml。此外,替考拉寧需在開始給藥時給予足夠的負荷劑量,再給予維持劑量,這樣可迅速達到較穩定的血藥谷濃度。因此,在臨床實踐中應合理監測其血藥濃度以保證療效。本研究采用內標法優化HPLC 法檢測血漿中替考拉寧的血藥濃度,旨在為臨床制定更合理的給藥方案提供借鑒。
1 材料與儀器
1.1 試藥
替考拉寧對照品(效價:894 U/mg,中國食品藥品檢定研究院,批號:130374-201002);哌拉西林鈉對照品(純度:98.0%,上海梯希愛化成工業發展有限公司,批號:5C8OB-CA);注射用替考拉寧[賽諾菲(北京)制藥有限公司,批號:CF20178,規格:0.2 g/支];乙腈、甲醇(色譜純,德國Merck 公司);二氯甲烷、磷酸、磷酸二氫鉀均為分析純(國藥集團化學試劑有限公司);實驗用水為Millipore 超純水。
1.2 儀器
1100 型高效液相色譜儀(美國Agilent 公司)(包括自動進樣器、真空脫氣機、四元泵、紫外檢測器、色譜工作站);Diamonsil C色譜柱(美國Agilent 公司,4.6 mm×150 mm,5 μm);VORTEX-5 型旋渦混合器(海門市其林貝爾儀器制造有限公司);Sorvall Stratos型高速離心機(德國Thermo 公司);BP211D 型電子分析天平(德國Sartorius 公司);GZ-1 型多用震蕩器(北京畢捷電機股份有限公司)。
2 方法與結果
2.1 HPLC 色譜條件
色譜柱:Diamonsil C柱(4.6 mm×150 mm,5 μm);流動相:乙腈-0.01 mol/L 磷酸二氫鉀溶液(25∶75)(用磷酸溶液調整pH 值為2.8);柱溫:30℃;流速:1.2 ml/min;進樣量:20 μl;檢測波長:215 nm。
2.2 溶液的配制方法
2.2.1 替考拉寧標準溶液 精密稱取替考拉寧對照品100.00 mg,置50 ml 容量瓶內,加入適量10%甲醇超聲溶解后稀釋至刻度,反復顛倒搖勻,經0.22 μm 微孔濾膜過濾,使成2.00 mg/ml 的標準溶液,置于-20℃冰箱冷凍保存。
2.2.2 內標溶液 精密稱取哌拉西林鈉對照品76.53 mg,置50 ml 容量瓶內,加超純水超聲溶解后稀釋至刻度,反復顛倒搖勻,得1.50 mg/ml 哌拉西林鈉內標儲備液。精密量取取儲備液1.0 ml,置10 ml容量瓶內,加超純水超聲溶解后稀釋至刻度,反復顛倒搖勻,經0.22 μm 微孔濾膜過濾,得150.00 μg/ml 內標溶液,置于-20℃冰箱冷凍保存。
2.2.3 血樣的處理方法 量取空白血漿、標準曲線血漿或質控血漿樣品400 μl 置1.5 ml 離心管內,加入50 μl“2.2.2”項下的內標溶液,再加入乙腈600 μl,渦旋震蕩2 min,離心半徑7 cm,離心轉速12 000 r/min,離心5 min 后得上清液。將上清液900 μl 移入另一個1.5 ml 離心管內,加入二氯甲烷400 μl 萃取,渦旋震蕩2 min,離心半徑7 cm,離心轉速12 000 r/min,離心5 min 后得上清液。經0.22 μm 微孔濾膜過濾,取上清液20 μl 進樣。
2.3 方法學考察
2.3.1 專屬性和系統適應性試驗 按“2.2.3”項處理血樣,按“2.1”項進樣分析,HPLC 圖譜見圖1。結果顯示,標準曲線血漿和質控血漿樣品的替考拉寧(取其主要組分TA2-2 吸收峰)、內標峰形均良好,保留時間分別為7.3 min 和12.5 min。因此該方法的專屬性良好。色譜柱的理論板數10 295,分離度均>1.5,拖尾因子分別為1.02 和1.03,符合《中華人民共和國藥典》的要求。

圖1 HPLC 色譜圖
2.3.2 標準曲線 吸取“2.2.1”項下的替考拉寧標準溶液50 μl,加入空白血漿950 μl,配制成濃度為100 μg/ml 樣品A;吸取A 溶液500 μl,加入空白血漿500 μl,配制成濃度為50 μg/ml 樣品B;依照同法繼續稀釋樣品,得25.000、12.500、6.250、3.125 μg/ml 系列濃度樣品C、D、E、F。按“2.2.3”項處理血樣,按“2.1”項進樣分析,以TA2-2 與內標峰面積比為橫坐標(X),質量濃度為縱坐標(Y,μg/ml),采用加權最小二乘法,得回歸方程:Y=11.295 386X-0.048 985(r=0.9987),在3.125~100.000 μg/ml 峰面積比與質量濃度呈良好的線性關系。
2.3.3 準確度 吸取“2.2.1”項下的替考拉寧標準溶液50 μl,加入空白血漿950 μl,配制成濃度為100 μg/ml樣品A;吸取A 溶液250 μl,加入空白血漿500 μl,配制成濃度為33.33 μg/ml 質控樣品H;依照同法繼續稀釋樣品,得濃度為11.11、3.70 μg/ml 質控樣品M、L,均平行制備5 份。按“2.2.3”項處理血樣,按“2.1”項進樣分析,每日各檢測1 次,共檢測3 d,計算批內及批間準確度。計算公式為:相對誤差(relative error,RE)=(實測值/標示值)×100%,見表1。結果顯示,低、中、高質量濃度質控樣品的批內及批間RE 均在85%~115%范圍內,表明分析方法的準確度良好。

表1 批內及批間準確度(%)
2.3.4 精密度 取“2.3.3”項下的質量濃度3.70、11.11、33.33 μg/ml 的低、中、高濃度質控樣品L、M、H。按“2.2.3”項方法處理血樣,按“2.1”項進樣分析,每日各檢測1 次,共檢測3 d,計算批內及批間相對標準差(relative standard deviation,RSD),見表2。結果顯示,低、中、高質量濃度質控樣品的批內及批間RSD>15%,表明分析方法的精密度良好。

表2 批內及批間精密度(%)
2.3.5 提取回收率 取 “2.3.3” 項下的質量濃度3.70、11.11、33.33 μg/ml 的低、中、高質量濃度質控樣品,測定得到的峰面積用Q 指代,取“2.2.1”項下的替考拉寧標準溶液加流動相稀釋的質量濃度3.70、11.11、33.33 μg/ml 的低、中、高質量濃度的替考拉寧溶液樣品,測定得到的峰面積用P 指代,均平行制備5 份。按“2.2.3”項方法處理血樣,按“2.1”項進樣分析,并計算提取回收率=(峰面積Q/峰面積P)×100%,見表3。結果顯示,低、中、高質量濃度樣品的替考拉寧和內標的提取回收率均在85%~115%范圍內,符合要求。
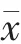
表3 提取回收率結果(%,±s,n=5)
2.4 樣品測定
5 例患者靜脈滴注替考拉寧0.4 g/次,1 次/d,在第2 天給藥前30 min 和第6 天給藥前30 min,采集靜脈血2 ml,采用含乙二胺四乙酸(ethylene diamine te traacetic acid,EDTA)抗凝劑的無菌采血管保存血樣。按“2.2.3”項方法處理血樣,按“2.1”項進樣分析,測定替考拉寧谷濃度,結果見表4。其中4 例患者的替考拉寧穩態谷濃度在10~25 μg/ml 有效谷濃度范圍內,另外1 例(編號2 患者)需及時調整劑量才能保證療效和用藥安全。

表4 5 例患者不同采血時機的替考拉寧谷濃度測定結果(μg/ml)
3 討論
3.1 色譜條件的選擇
與甲醇-水流動相比較,乙腈-水流動相可獲得更好的峰型和分離度。當乙腈與水體積比為25∶75 時,替考拉寧與內標的保留時間分別為7.3、12.5 min,且此方法的分離度均>1.5,能滿足基線分析的要求。在流動相內添加不同的離子對試劑可進一步改善分離效果和峰型,如:乙酸銨、甲酸銨、磷酸二氫鉀等。結果表明,在水中加入0.01 mol/L 磷酸二氫鉀時峰型較好。流動相的pH 值能影響替考拉寧和內標的保留時間,當pH 值較高時替考拉寧的保留時間較長,而內標的保留時間較短,因此選擇合適的pH 值為2.8。本研究最終選擇流動相為乙腈-0.01 mol/L 磷酸二氫鉀溶液(25∶75)(用磷酸溶液調整pH 值為2.8)。
替考拉寧在215 nm 和240 nm 均有較強的紫外吸收峰,選擇以上兩種波長進行考察。結果發現檢測波長為215 nm 時,替考拉寧和內標的峰形良好,血漿雜質吸收較弱,對測定替考拉寧影響較小;檢測波長為240 nm 時,血漿雜質吸收雖弱,但替考拉寧的出峰較小,峰面積不如215 nm,此波長的檢測限和定量限不如215 nm 好。因此,本研究選擇215 nm 為最佳的檢測波長。
3.2 內標的選擇
內標法檢測替考拉寧血藥濃度的內標可選擇對羥基苯甲酸甲酯、哌拉西林鈉、苯巴比妥、香草醛、拉莫三嗪等。本研究除哌拉西林鈉外,其他內標的保留時間均較短,容易和血漿樣品中的其他物質混合,影響檢測結果,因此選擇哌拉西林鈉為內標。但由于臨床中可能會有同時使用替考拉寧與哌拉西拉鈉的患者,這樣的血樣可以用外標法檢測。本研究經方法學驗證,外標法也可以滿足方法學的相關要求。
3.3 血漿樣品前處理溶劑的選擇
替考拉寧穩定性雖好但極性較大,因此要選擇合適的萃取試劑。血漿樣本先采用乙腈沉淀蛋白后,本研究考察不同有機溶劑萃取的效果,有甲基叔丁基醚和二氯甲烷等,發現用二氯甲烷萃取樣品的分離度比甲基叔丁基醚更優。所以本研究最終選擇二氯甲烷作為液-液萃取有機溶劑。
綜上所述,采用HPLC 法測定血漿中替考拉寧的血藥濃度,方法簡便可行,專屬性強,標準曲線峰面積比與質量濃度呈良好的線性關系,重現性好,準確度、精密度、提取回收率等方法學指標均符合《中華人民共和國藥典》要求。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
云南醫藥(2019年3期)2019-07-25 07:25:14
海南醫學(2016年8期)2016-06-08 05:43:00
海峽科技與產業(2016年3期)2016-05-17 04:32:12
