單臂試驗藥物醫(yī)保準入評估的方法學思考
2022-08-30 09:43:20馮依曼丁錦希
中國醫(yī)療保險 2022年8期
關(guān)鍵詞:療效
馮依曼 丁錦希,2 李 偉,2 方 剛
(1中國藥科大學國際醫(yī)藥商學院 南京 211198;2中國藥科大學醫(yī)藥市場準入政策研究中心 南京 211198)
1 需求與挑戰(zhàn)
1.1 單臂試驗藥物
單臂試驗藥物是指某藥物上市注冊審評的關(guān)鍵臨床試驗采用單臂試驗方案。而單臂試驗(single-arm trial, SAT)是指臨床試驗方案中不設立平行對照組,而采用外部對照(如歷史對照),將接受新藥治療的一組患者與該研究以外一組患者的臨床結(jié)果進行比較的臨床研究。
與隨機對照試驗(randomized controlled trial, RCT)不同,單臂試驗并非將受試者隨機分為兩組,而是將所有受試者納入一組,且都予以新藥治療。
1.2 醫(yī)保準入需求
單臂試驗藥物通常治療嚴重疾病、臨床急需且療效顯著,往往通過附條件審批方式快速上市。隨著更多的單臂試驗藥物加速上市,其醫(yī)保準入需求顯著增加。全球范圍內(nèi),針對單臂試驗藥物醫(yī)保準入而出具的衛(wèi)生技術(shù)評估報告數(shù)量快速上升,2019年(102份)是2011年(8份)的近13倍[1]。
近年來,我國單臂試驗藥物醫(yī)保準入需求呈幾何指數(shù)增長。2017年—2021年,我國醫(yī)保準入18個單臂試驗藥物/適應癥,絕大多數(shù)藥物治療復發(fā)難治腫瘤。2021年7月醫(yī)保目錄調(diào)整時,近一年內(nèi)上市的14個單臂試驗藥物/適應癥全都申報醫(yī)保準入,但成功率僅為64%。預計2022年至少17個單臂試驗藥物/適應癥存在醫(yī)保準入需求。
1.3 準入評估挑戰(zhàn)
單臂試驗藥物主要面臨以下四點準入評估挑戰(zhàn)。
一是無參照藥物。上市注冊臨床試驗時,單臂試驗方案僅設置試驗組,未設置對照組。故醫(yī)保準入評估時的相對療效評估和成本效果分析較難選擇參照藥物。
二是無直接比對數(shù)據(jù)。單臂試驗只有試驗組患者一組單獨數(shù)據(jù),缺失與對照藥物的療效直接比對數(shù)據(jù),難精準評估其真實臨床價值。
三是測量指標為替代終點。單臂試驗的臨床結(jié)果測量指標通常為替代終點而非臨床終點,替代終點結(jié)果并不一定能詮釋患者的長期健康結(jié)局。
四是證據(jù)不穩(wěn)定。未采取隨機化和盲法、隨訪時間短、患者樣本量小等缺陷導致單臂試驗藥物的證據(jù)質(zhì)量相對較差,較難獲得穩(wěn)健的評估結(jié)果。
本文基于單臂試驗藥物上述挑戰(zhàn),研究針對性解決方案,探討如何通過優(yōu)化評估方法,控制評估結(jié)果不確定性,以科學評判單臂試驗藥物的有效性和經(jīng)濟性,為醫(yī)保準入決策提供精準參考。
2 無參照藥物
2.1 是否選擇參照藥物
單臂試驗藥物的臨床試驗方案未設立對照組,通常上市注冊審評時無對照藥物。這可能導致理解誤區(qū),即醫(yī)保準入時也難以選擇參照藥物,應該選擇空白對照。
但事實并非如此。筆者比對國家醫(yī)保局公開的2021年醫(yī)保藥品目錄調(diào)整形式審查材料和目錄調(diào)整結(jié)果后發(fā)現(xiàn),形式審查材料中選擇了明確參照藥物的單臂試驗藥物,其準入成功率明顯更高。近一年內(nèi)上市的14個單臂試驗藥物/適應癥中,9個藥物/適應癥提交了參照藥物,都成功準入(見表1)。而另5個單臂試驗藥物/適應癥未填寫參照藥物,選擇空白對照的,其準入結(jié)果卻都不理想。可見,是否提交參照藥物與準入結(jié)果密切相關(guān),應積極申報并提交陽性參照藥物。

表1 2021年成功準入的單臂試驗藥物的參照藥物選擇情況
那么,為什么單臂試驗藥物在上市審評試驗時無對照藥物,而醫(yī)保準入時,卻可以選擇陽性參照藥物?
其主要原因是:新藥上市審評和醫(yī)保準入之間存在較長的時間差。在單臂試驗藥物設計確定臨床試驗方案時,其同適應癥、同靶點、同機制藥物往往也在臨床試驗或上市審評過程中,所以只能選擇無對照藥物。但在其漫長的臨床試驗和上市審評期間,其同適應癥、同機制、同靶點藥物已上市且準入醫(yī)保,所以在醫(yī)保準入時就可以選擇陽性參照藥物。
以帕米帕利為例,它是治療晚期卵巢癌、輸卵管癌或原發(fā)性腹膜癌的多腺苷二磷酸核糖聚合酶抑制劑(poly ADP-ribose polymerase,PARP抑制劑)。帕米帕利的關(guān)鍵臨床試驗于2016年12月開啟,當時雖然同為PARP抑制劑的奧拉帕利臨床研究進程快于帕米帕利,但尚未注冊上市。因缺乏臨床實踐公認的藥物可作為陽性對照,且考慮到倫理道德等原因,未采用安慰劑對照,故帕米帕利采取了單臂試驗方案。
但帕米帕利在2021年4月上市時,奧拉帕利已于2018年在我國批準上市,并于2019年準入我國醫(yī)保藥品目錄。故2021年6月,帕米帕利在醫(yī)保準入申報書中以奧拉帕利為陽性參照藥物,并成功準入醫(yī)保藥品目錄。
2.2 如何選擇參照藥物
2.2.1 代際相近。首先,選擇代際相近的藥物為參照藥物,尤其是同機制藥物。按同疾病領(lǐng)域內(nèi)取得的歷次突破性進展類型進行排序,形成代際順序。
如腫瘤領(lǐng)域藥物主要分為三個代際[2],第一代為細胞毒性的化療藥物,如達卡巴嗪;第二代為靶向治療的小分子化藥和生物制品,如曲妥珠單抗;第三代為免疫療法藥物,如嵌合抗原受體T細胞(chimeric antigen receptor T-cell, CAR-T)。隨著創(chuàng)新能力提高,新代際藥物的有效性和安全性逐漸提升,代際相近的藥物可比性更高。
加拿大藥品與衛(wèi)生技術(shù)局(Canadian Agency for Drugs and Technologies for Health,CADTH)準入審評CAR-T產(chǎn)品阿基侖賽(Yescarta)時,以同為3代免疫療法的另一款CAR-T產(chǎn)品Kymriah為參照藥物[3]。
2.2.2 序貫相近。其次,選擇臨床治療序貫相近的藥物為參照方案,使得二者治療人群特征、治療難度和愈后效果接近,可比性更強。
根據(jù)公開的形式審查材料,2021年我國醫(yī)保準入審評維迪西妥單抗時,目錄內(nèi)無該治療領(lǐng)域同代際、同機制藥物,所以企業(yè)選擇了序貫相同的阿帕替尼作為參照藥物。根據(jù)《中華醫(yī)學會胃癌臨床診療指南(2021版)》,維迪西妥單抗和阿帕替尼均被推薦用于三線的晚期轉(zhuǎn)移性胃癌[4]。符合序貫相近原則,故被國家醫(yī)保局批準,并成功準入醫(yī)保。
2.2.3 治療方案。若上述思路均無合適參照藥物,可考慮選擇治療方案為參照。從2021年起,我國醫(yī)保藥品目錄調(diào)整工作方案允許臨床“治療方案”為談判新藥的參照方案,這為first in class藥物選擇參照治療方案奠定了制度基礎[5]。
以依庫珠單抗為例,作為罕見病非典型溶血性尿毒綜合征(atypical hemolytic uremic syndrome,aHUS)在全球范圍內(nèi)的唯一治療藥物,其在英國[6]和法國[7]的準入審評中均采用最佳支持療法作為參照方案,即血漿置換治療。根據(jù)我國《罕見病診療指南(2019年版)》,在依庫珠單抗應用于治療aHUS之前,血漿置換是aHUS的一線治療方案,因此,可以考慮以血漿置換治療方案作為依庫珠單抗的參照方案。
3 無直接比對數(shù)據(jù)
3.1 療效比較方法
醫(yī)保準入評估主要是通過新藥(談判藥)與參照藥之間的相對療效比較,以確定新藥為參保人帶來臨床獲益的增量值[8]。如圖1所示,主要比較方法有以下三種:陽性對照頭對頭試驗的直接比較、傳統(tǒng)RCT以安慰劑為錨點的間接比較和單臂試驗無錨點的間接比較。以參照藥為陽性對照組的頭對頭臨床試驗(圖1.A)是最理想的療效比較數(shù)據(jù)來源,兩組患者在同一個試驗背景下,患者同質(zhì)、評估方法相同,得到的療效比較結(jié)果受其他因素干擾最小,最可靠。但是由于成本更高、失敗風險較大等原因,藥品臨床試驗設計中陽性對照的頭對頭試驗數(shù)量較少,更多的是談判藥與安慰劑對比。
因此,當談判藥與參照藥的RCT都是與安慰劑對比時,兩者之間的比較常以安慰劑為錨點或參照系,使用統(tǒng)計學方法間接比較相對療效(圖1.B)。該方法雖然不如頭對頭臨床試驗比較結(jié)果的精準度高,卻是目前常見的、通用的方法。
然而,單臂試驗缺乏安慰劑組作為錨點(圖1.C),此類談判藥與參照藥之間的療效比較困難重重。
3.2 如何科學比對療效
針對單臂試驗藥物的相對療效評估,最方便、快捷的方法是簡單比較(nave comparison),即將談判藥組數(shù)據(jù)與參照藥組兩個不同臨床試驗數(shù)據(jù)直接比對。但簡單比較法的局限性明顯,兩個試驗間樣本人群異質(zhì)性,很可能導致比較結(jié)果存在較大偏差[9]。
目前,國際上多采用匹配調(diào)整間接比較法(matching-adjusted indirect comparison, MAIC)評估單臂試驗藥物與參照藥的相對療效,即將兩個試驗的受試患者的基線條件調(diào)至一致后,再比較療效。
如圖2所示,匹配調(diào)整間接比較的前提是,能夠獲取談判藥單臂試驗A的患者個體數(shù)據(jù)和參照藥臨床試驗B的公開匯總數(shù)據(jù)。可分為四個步驟[10]:一是通過系統(tǒng)文獻綜述,選擇與單臂試驗A納排標準和基線特征相近的參照藥臨床試驗B;二是選取兩個臨床研究方案共有的患者基線特征(年齡、性別、人種、疾病嚴重程度等)和臨床結(jié)局指標,用于跨試驗匹配和比較;三是調(diào)整匹配兩組人群的基線特征,加權(quán)療效數(shù)據(jù)。將滿足試驗A納排標準,但不滿足試驗B納排標準的患者數(shù)據(jù)刪去,再將試驗A患者按照其參加試驗B的概率重新加權(quán);四是比較匹配調(diào)整后的療效結(jié)果。
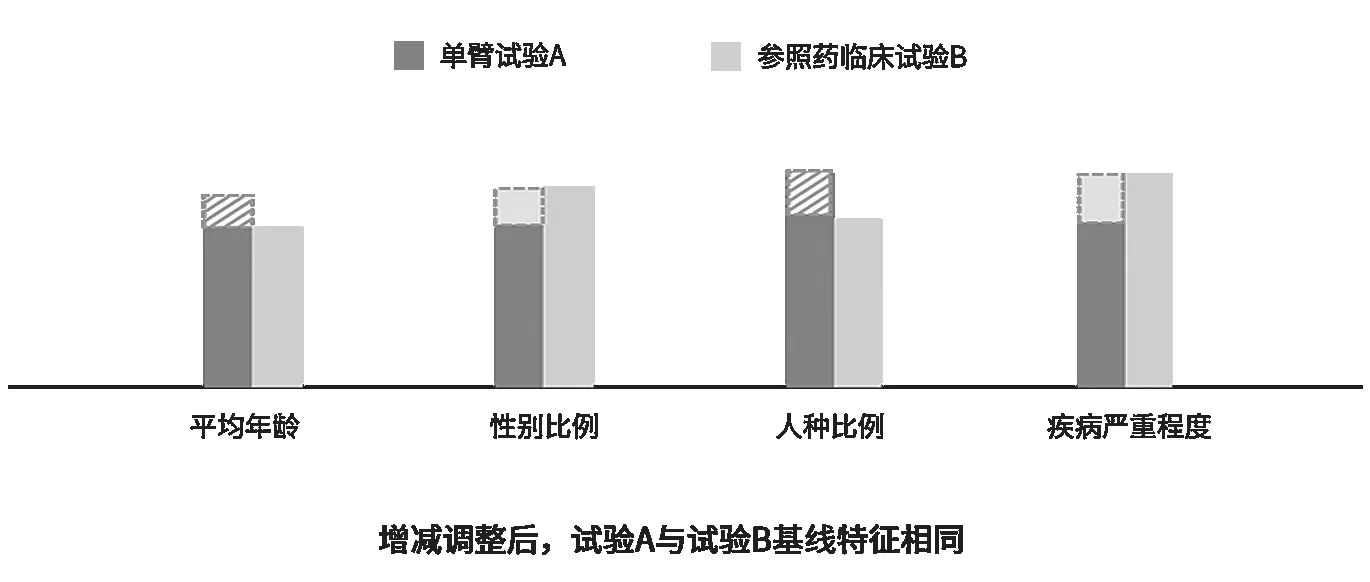
圖2 匹配調(diào)整間接比較的患者基線特征示意圖
英國單臂試驗藥物醫(yī)保準入評估中,匹配調(diào)整間接比較方法應用較為成熟。經(jīng)筆者統(tǒng)計,NICE截至2022年3月31日發(fā)布的所有46份單臂試驗藥物的評估報告中,采用匹配調(diào)整間接比較的報告數(shù)量最多,為28份(61%)。NICE還專門發(fā)布了相關(guān)技術(shù)指導文件《Populationadjusted indirect comparisons(MAIC and STC)》[11]。其成功經(jīng)驗值得我們借鑒。
需要注意是,匹配調(diào)整間接比較方法中最關(guān)鍵的步驟是第三步,即如何調(diào)整單臂試驗A患者基線特征。以治療基因型3丙肝的藥物評估為例[12],達拉他韋+索非布韋是待評估聯(lián)合治療方案,索非布韋+利巴韋林是參照方案。提取兩組方案人群的基線特征之后,采取傾向性評分加權(quán)的統(tǒng)計學方法進行調(diào)整,使評估方案的基線特征與參照方案相同,如將評估方案中白人患者比例從90.3%調(diào)至96.4%(見圖3)。

圖3 匹配調(diào)整間接比較的案例
人群基線均衡后,療效可比性提高。評估方案的治療終點后12個月隨訪時,HCV-RNA陰性(SVR12)比例從89.6%降至88.8%,對比參照方案的療效優(yōu)勢縮小[12]。
4 替代終點
4.1 測量指標和時間
藥物臨床試驗通常采用一定療效終點指標來衡量患者的臨床獲益,包括臨床終點和替代終點這兩類指標。在抗腫瘤藥物的臨床試驗中,多以總生存期(overall survival,OS)這一臨床終點為主要指標,OS定義明確且客觀穩(wěn)健,能直接反映患者生存獲益。
但是,單臂試驗藥物隨訪時間較短,多采用替代終點間接反映臨床獲益,例如客觀緩解率(objective response rate, ORR)。雖然使用替代終點能夠降低試驗成本、加快藥品上市,但也存在療效不確定性大、與臨床終點的可替代性尚不明確等問題。
與RCT相比,單臂試驗的隨訪期較短。有學者統(tǒng)計了2010年至2020年發(fā)表在主流期刊的腫瘤領(lǐng)域RCT,其平均中位隨訪時間為25個月[13]。而單臂試驗隨訪期多為14個月左右(根據(jù)2021年我國醫(yī)保準入成功的單臂試驗藥物的申請上市技術(shù)審評報告和說明書總結(jié)),顯著低于RCT。在準入審評時,以短期試驗數(shù)據(jù)推算長期臨床獲益的不確定性較大,患者實際獲益情況仍不明確。
此外,相關(guān)研究結(jié)果顯示,替代終點與臨床終點的相關(guān)性較弱。有學者系統(tǒng)綜述了腫瘤治療領(lǐng)域內(nèi)所有報告了OS與替代終點關(guān)系的研究文獻,僅有11篇(12%)報告了高度相關(guān)性,9篇(10%)報告了中度相關(guān)性,34篇(38%)報告了低度相關(guān)性[14]。替代終點與臨床終點的弱相關(guān)性導致藥物真實療效的不確定性,加大了醫(yī)保準入的決策風險。
4.2 如何評估長期臨床獲益
4.2.1 增加隨訪時間,補充終點指標。我國《藥品附條件批準上市技術(shù)指導原則(試行)》規(guī)定,單臂試驗藥物在使用替代終點獲得附條件批準上市后,應在規(guī)定期限內(nèi)設計并完成以臨床終點為主要終點指標的確證性臨床試驗。一般是隨機對照確證性研究,進一步提供有效性數(shù)據(jù),證實該治療給患者帶來的生存獲益,以獲得完全批準。
原單臂試驗的繼續(xù)隨訪和開展確證性RCT,均可獲得豐富的臨床試驗數(shù)據(jù)。新藥醫(yī)保準入時,往往已經(jīng)得到比上市審批時觀測時間更長久、測量結(jié)果更成熟的試驗數(shù)據(jù),可以供醫(yī)保準入決策參考。
以治療復發(fā)或難治性B細胞急性淋巴細胞白血病的CAR-T藥物Kymriah為例,其基于24個月隨訪時間獲得的臨床數(shù)據(jù)批準上市,而英國NICE基于36個月隨訪時間獲得的同一臨床試驗的擴展數(shù)據(jù),對Kymriah進行綜合審評并納入報銷范圍[3]。
我國國家醫(yī)保局應加強與國家藥監(jiān)局的銜接聯(lián)動,將完成確證性臨床試驗轉(zhuǎn)為完全批準作為單臂試驗藥物醫(yī)保準入的申報前提,等待臨床終點數(shù)據(jù)充分成熟后再準入醫(yī)保。或者,針對臨床急需的單臂試驗藥物,亦可要求企業(yè)準入申報時提供最新的臨床試驗隨訪數(shù)據(jù)和療效結(jié)果,以及真實世界研究等所有相關(guān)臨床數(shù)據(jù),以控制醫(yī)保準入評估的不確定性。
4.2.2 選取合適模型,擬合生存曲線。對于臨床需求迫切,醫(yī)保準入周期短的單臂試驗藥物進行準入評價時,就需要選取合適外推模型。根據(jù)短期臨床試驗匯報的生存曲線,構(gòu)建合理的分布函數(shù)對生存曲線進行外推,以模擬患者長期的生存情況。
采用不同分布函數(shù)模擬的長期生存結(jié)果可能具有顯著差異,對于臨床療效本就不確定的單臂試驗藥物,外推模型的選取更加關(guān)鍵。
英國NICE審評治療黑色素瘤的伊匹單抗時,根據(jù)單臂試驗CA184-024的5年隨訪數(shù)據(jù)發(fā)現(xiàn),采用分段模型擬合的生存結(jié)果與實際臨床研究結(jié)果吻合度較高。而混合治愈模型、標準參數(shù)模型、樣條曲線模型的擬合效果較差,低估了5年生存率(見表2)[15]。
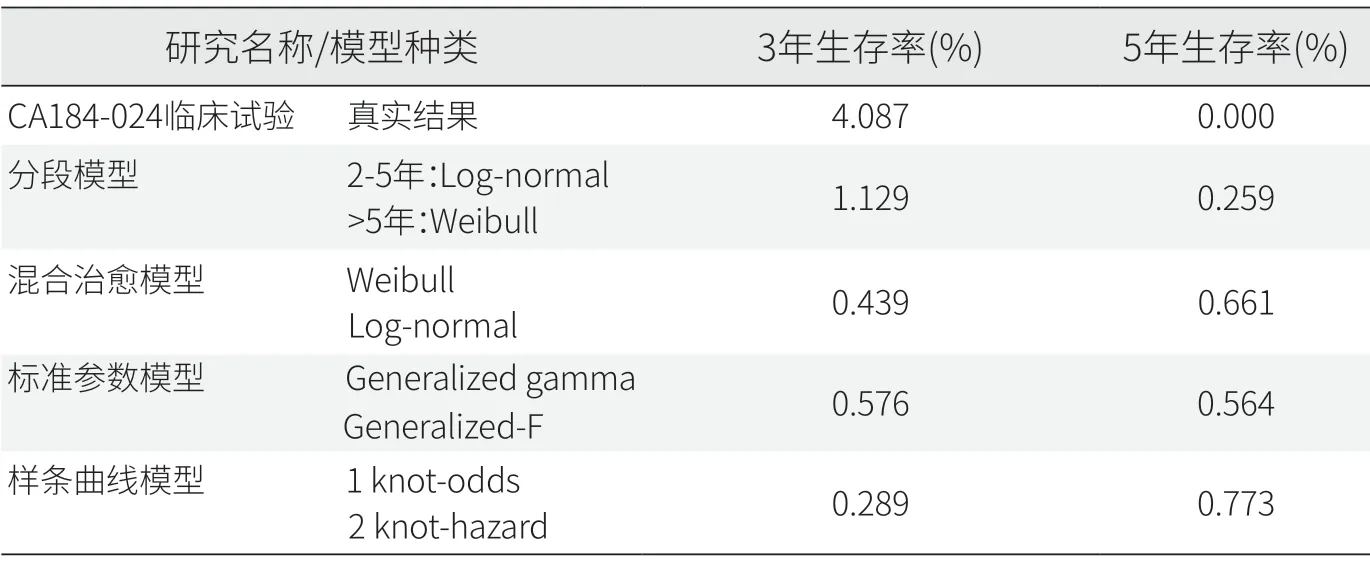
表2 伊匹單抗+達卡巴嗪實際研究與外推模型的長期生存數(shù)據(jù)對比
因此,需要綜合考慮疾病臨床特征、患者人群情況、藥物特性等因素,采用適宜的外推模型擬合生存曲線,盡量準確測定長期生存率等關(guān)鍵準入評估數(shù)據(jù)。
以CAR-T產(chǎn)品為例,推薦使用混合治愈模型。其藥物特征是能使部分患者實現(xiàn)長期緩解[16],治療后終點事件發(fā)生概率低,表現(xiàn)為“L”形的Kaplan-Meier(K-M)生存曲線(見圖4)。混合治愈模型將研究人群分為治愈患者和未治愈患者,更貼合CAR-T臨床實際情況,為成本效果分析提供了更準確的長期療效數(shù)據(jù)[17]。
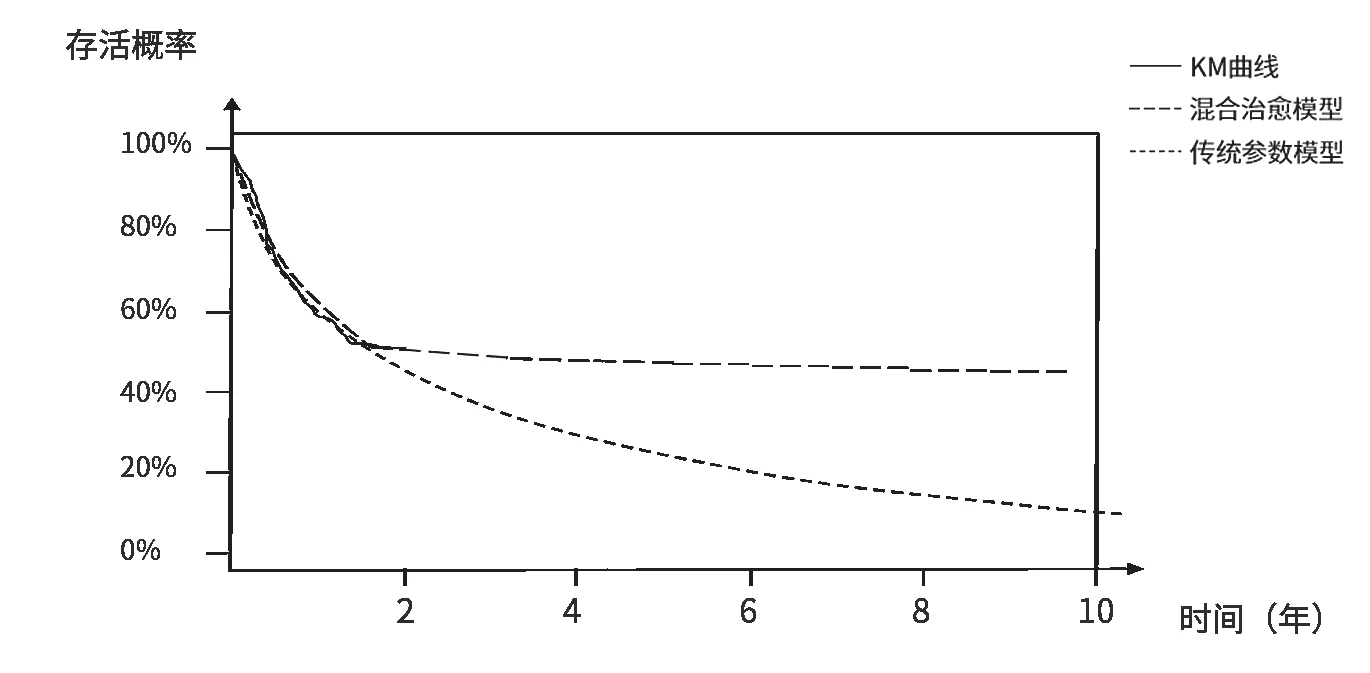
圖4 不同模型擬合外推CAR-T產(chǎn)品生存曲線示例
5 證據(jù)不穩(wěn)定
5.1 成本效果增量比不確定性大
各國衛(wèi)生技術(shù)評估機構(gòu)普遍推薦RCT作為醫(yī)保準入評估的主要審評證據(jù)。而單臂試驗位于循證醫(yī)學證據(jù)金字塔的下方,其療效結(jié)果存疑、證據(jù)質(zhì)量較低[18]。
單臂試驗有效性證據(jù)質(zhì)量較低,主要原因有三點:一是未設立對照組,較難將治療效果與安慰劑效果、疾病自然史結(jié)果區(qū)分開來[19];二是單臂試驗不涉及隨機與盲法、試驗組與外部對照的人群基線存在差異,較難排除混雜因素對結(jié)果的影響[20];三是使用替代臨床終點間接反映臨床獲益[21],且隨訪時間較短、入組人數(shù)較少,存在試驗結(jié)果誤導風險。
體現(xiàn)藥物有效性的療效參數(shù)是藥物經(jīng)濟學測算的最重要參數(shù)。但是,由于單臂試驗證據(jù)質(zhì)量較低,以及在療效比較中MAIC等方法的不穩(wěn)定性,導致藥物經(jīng)濟學測算中療效關(guān)鍵參數(shù)估算不確定性較大,進而導致增量成本效果比(incremental cost effectiveness ratio,ICER)測算結(jié)果的變化范圍較大,結(jié)果穩(wěn)健性低。
5.2 如何減少不確定性影響
5.2.1 強化敏感性分析。藥物經(jīng)濟學的敏感性分析結(jié)果與基礎分析結(jié)果同樣重要,甚至更加重要,單臂試驗藥物需要更加重視敏感性分析。為避免潛在偏倚,應當盡量將所有參數(shù)和假設列入分析備選項,根據(jù)參數(shù)估計值的95%置信區(qū)間、高值與低值、文獻情況以及真實世界情況,確立參數(shù)的變動范圍以及分布[22],如不良反應發(fā)生率的95%置信區(qū)間。
5.2.2 調(diào)整敏感參數(shù)。在敏感性分析中,應關(guān)注能顯著影響結(jié)果甚至翻轉(zhuǎn)成本效果結(jié)論的敏感參數(shù),選出這些參數(shù)并展開重點分析。有研究顯示,導致抗癌藥決策不確定性最高的因素包括,生存推斷、健康效用值、藥品/護理/不良反應成本[23]。單臂試驗藥物在敏感性分析中應注重上述參數(shù)。
重點分析分為兩步,先評估這些參數(shù)在真實世界的情況,詳盡收集臨床患者資料,將參數(shù)的取值修正為真實世界臨床實踐的確定值或變化范圍;然后根據(jù)調(diào)整后的參數(shù)計算得出新ICER值,同時報告調(diào)整前后的參數(shù)和對應的ICER值,解釋參數(shù)變化的原因,以及ICER值變動對決策的意義。
以治療大B細胞淋巴瘤的CAR-T藥物Yescarta為例,成本效果分析中的單因素敏感性分析顯示,對ICER影響最大的因素是患者平均年齡。若根據(jù)臨床試驗ZUMA-1數(shù)據(jù),患者基線平均年齡為58歲,ICER值為17.03萬美元/QALY;若根據(jù)真實世界數(shù)據(jù),患者年齡調(diào)整為67歲,ICER值將增加至20.81萬美元/QALY。鑒于此,加拿大CADTH評估時將患者年齡基線調(diào)整為67歲,以減少不確定性影響,進行更為準確的評估[24]。
6 小結(jié)
隨著醫(yī)藥創(chuàng)新投入加速增長、藥品監(jiān)管部門審評機制不斷優(yōu)化,大量單臂試驗藥物批準上市。如何提高醫(yī)保準入評估的準確度,在醫(yī)保基金可持續(xù)性與創(chuàng)新藥物可負擔性之間實現(xiàn)有效平衡,是國家醫(yī)保目錄管理面臨的新課題。
本文針對單臂試驗藥物的準入難點,結(jié)合國內(nèi)外的實踐經(jīng)驗,提出了優(yōu)化思路,以期實現(xiàn)精準評估,真實體現(xiàn)單臂試驗藥物的臨床價值。盡管如此,不確定性較大的問題依然存在。因此,今后仍需探索創(chuàng)新支付,根據(jù)準入后真實世界中的療效和患者人群予以不同基金支付金額。通過“精準評估+創(chuàng)新支付”的綜合治理模式,實現(xiàn)醫(yī)保基金效率最大化,滿足廣大患者對醫(yī)療新技術(shù)不斷增長的需求。
猜你喜歡
中華養(yǎng)生保健(2020年1期)2020-11-16 00:47:38
中國中醫(yī)急癥(2019年10期)2019-05-21 07:20:30
中國醫(yī)藥指南(2017年3期)2017-11-13 02:57:31
中國民族醫(yī)藥雜志(2016年9期)2016-05-09 07:51:44
中國民族醫(yī)藥雜志(2016年9期)2016-05-09 07:51:44
中國衛(wèi)生標準管理(2015年8期)2016-01-15 03:58:45
中國康復理論與實踐(2015年10期)2015-12-24 05:42:46
中國衛(wèi)生標準管理(2015年13期)2015-01-26 21:05:38
中國醫(yī)學科學院學報(2014年6期)2014-03-11 20:26:14
中國中醫(yī)藥現(xiàn)代遠程教育(2014年21期)2014-03-01 04:32:10
