基于圖神經網絡的藥物相互預測方法
2022-08-31 19:18:39馬怡青蔡美玲陳明成明俊
電腦知識與技術 2022年18期
關鍵詞:深度學習
馬怡青 蔡美玲 陳明 成明俊


摘要:藥物-藥物相互作用(Drug-drug interactions, DDIs)指病人在一定時間內服用兩種及以上藥物后藥物產生的復合效應,可表現為藥性增強或減弱。本文提出一種基于圖神經網絡模型的預測方法,在已有藥物間相互作用基礎上,結合藥物化學結構特征等屬性,分進行藥物間相互作用預測實驗。
關鍵詞:藥物相互預測;圖神經網絡;符號網絡;深度學習
中圖分類號:TP391? ? ? 文獻標識碼:A
文章編號:1009-3044(2022)18-0061-03
開放科學(資源服務)標識碼(OSID):
1 引言
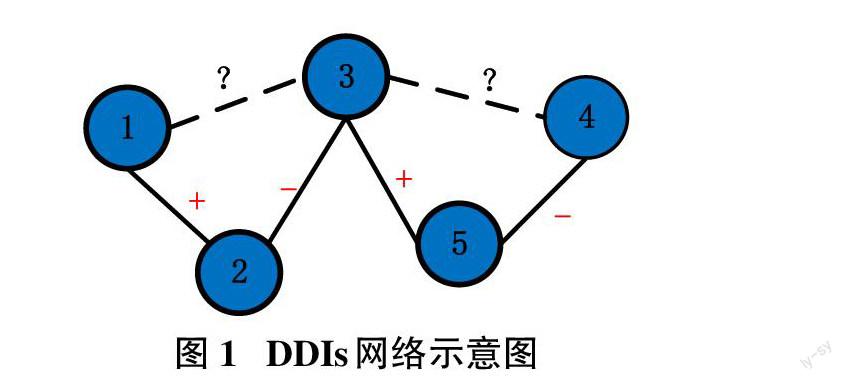
藥物-藥物相互作用(Drug-drug interactions, DDIs)指的是兩種及以上藥物混合使用時藥物產生的副作用效果[1],具體可表現為藥效增強或藥效減弱作用。這種現象在現實用藥中表現為藥性、毒性等增強或減弱。如果將藥物間的增強關系視為是正向作用,減弱關系視為是負向作用,那么藥物間相互作用關系可以使用一張圖的鄰接表進行表示。
假定使用圖[G(D,E)]來表示一個DDIs網絡,其中[D=d1,d2,...,dm]集合, 表示大小為[m]的藥物結點[d]集合;[E]表示藥物結間關系,如圖1所示。圖中,結點1和結點2間存在已證明的增強關系;結點2和結點3間存在已證明的減弱關系,而結點1和結點3間的關系是未知。通常,圖[G(D,E)]可以用一個[m×m]的鄰接對稱矩陣[Am×m=aij]表示,如式(1)所示,1表示藥物結點間存在增強關系,-1則為減弱關系,0則為未知。預測實驗是根據此鄰接矩陣中的非0元素信息,對0值元素預測。
[A=0100010-1000-10010000-1001-10]? ? ? ? ? ? ? ? ? ? ? ? ? (1)
2 相關工作
對于藥物間相互作用預測,生物實驗雖然準確,但成本高、耗時久。計算方法相反,所以在工業界和學術界取得很大關注。多數現有的基于深度學習的預測方法,主要為傳統的二值預測,即預測某一藥物對間產生DDIs的可能性。Liu等人利用隨機森林預測模型結合多特征進行藥物相互作用預測[2];Liu等人基于膠囊網絡的動態路由機制對文本中的藥物相互作用關系進行抽取[3];Feng等人結合圖卷積網絡GCN和深度神經網絡DNN進行藥物相互預測[4]。考慮DDIs更具體的增強或減弱作用,必須綜合考慮DDIs網絡結構。本文通過使用流行的圖神經網絡模型—SGCN模型,在式(1)定義的符號網絡上進行藥物間相互作用預測。
3 基于結點嵌入的DDIs預測框架
結點嵌入的目標是對結點進行編碼,生成一種結點的特征向量表示,這種向量表示不僅應體現結點的自身屬性,同時應能反應圖中結點間鄰居關系。考慮到已知的藥物間相互作用關系和藥物自身的特征表示,本文分兩步驟來解決DDIs預測問題,如圖2所示:
Step1特征提取:針對給定的鄰接對稱矩陣[A]和對應結點的特征矩陣[T],通過圖神經網絡模型,針對所有藥物結點得到一種低維向量表示[Z],[F1=A,T→Z]。
Step2相互作用預測:對任何一對藥物結點對[di]和[dj],將它們的低維向量表示[Zi]和[Zj][Zi,Zj∈Z]進行拼接作為藥物節點對的特征表示,使用線性分類器對[di]和[dj]的相互作用關系進行類別預測,[F2:(Zi,Zj)→0,1,-1]。
本文使用線性回歸模型對藥物間相互關系類型的進行預測。對藥物節點對的特征表示,通過線性分類器將它映射到3種情況{0,1,-1},保留正負情況中較大概率一方作為結果。
3.1 基于SGCN的結點嵌入
傳統的圖卷積神經網絡例如GCN[5],只考慮結點間是否有邊,即鄰接矩陣元素只考慮0或1,不適用于符號預測。
SGCN理論[6]出自平衡理論。平衡理論認為“朋友的朋友是朋友”而“朋友的敵人是敵人”。假設從結點[i]出發,經過[l]條邊可以抵達結點[j],若此路徑包含偶數條負邊,則[j∈Bi(l)],即結點[j]屬于結點[i]的[l]層朋友集合;若此路徑包含奇數條負邊,則[j∈Ui(l)],即結點[j]屬于結點[i]的[l]層敵人集合。
假設[h0i]是結點[i]的原始特征,維度大小為[din]。SGCN模型中結點[i]第一層隱藏層輸出分別考慮它的直接朋友和直接敵人的特征表示,定義如式(2):
[hB(1)i=σ(WB(1)[j∈N+ih(0)jN+i,h(0)i]),hU(1)i=σ(WU(1)[k∈N-ih(0)kN-i,h(0)i])] (2)
其中[σ]是一個非線性激活函數,在實現中選用ReLu函數。[WB1,WU1∈Rdoutx2din]是模型分別針對結點[i]的直接朋友集合[Bi(1)]和直接敵人集合[Ui(1)]的變換矩陣,[dout]是隱藏層輸出向量的長度。
結點[i]的第[l]層表達以迭代方式同時考慮[l-1]層鄰結點的朋友集合、敵人集合和自身表達。聚合公式如式(3):
[hB(l)i=σ(WB(l)[j∈N+ihB(l-1)jN+i,k∈N-ihU(l-1)kN-i, hB(l-1)i]), ]
[hU(l)i=σ(WU(l)[j∈N+ihU(l-1)jN+i,k∈N-ihB(l-1)kN-i,hU(l-1)i])]? ? ? (3)
其中[ WBl,WUl∈Rdoutx3din]。
經過兩次聚合,可以獲取藥物結點[i]的兩個隱藏層表示,稱為[hB(2)i]和[hU(2)i],這里將這兩個隱藏層輸出進行拼接作為結點
4 實驗
4.1實驗數據集
本文使用了來自四篇DDIs預測相關性的論文中提供的不同數據,包括4個數據集,如表1所示。其中, DB1有603個藥物節點[1]; DB2有568個藥物節點[7]; DB3有1562個藥物節點,附有881維的化學結構特征和1642維的藥物捆綁蛋白質結構特征[8],都是二值信息; DB4[4]有1934個藥物節點。
4.2實驗設置
本文選取了通用的評價指標來對本文提出的算法進行性能評估,包括精確度(accuracy)、準確率(precision)、召回率(recall)、[F1]指標和AP指標,同時還計算AUC和AUPR值。
以上指標中,有一部分計算結果是針對模型的預測得分。本文考慮兩種角度的預測打分,一類是根據線性分類器得到的預測概率進行計算打分(連續值),一類是將對線性分類器得到的預測概率進行處理,選擇正負邊中概率較大的一方作為最終預測結果作為分數。
5 實驗結果
5.1 SGCN參數調諧
參數設置上,本文使用格點搜索的方法,尋找最優指標對應的參數。降低后的維度分別為:8、16、32、64、128、256。
綜合參數調諧結果,本文后續實驗主要基于DB3進行,SGCN最優的參數設置:降低后的維度為256時,實驗結果相較于其他參數設置,表現出更好的綜合指標結果。
5.2 藥物化學結構特征,藥物蛋白質結構特征等特征比較
實驗基于DB3數據集進行參數比較和指標評估。藥物的特征的包括:藥物的化學結構特征、藥物的蛋白質屬性特征以及基于SSE方法生成的藥物在網絡中產生的結構特征[6]。
實驗中發現藥物化學結構PubChem特征單獨使用時,指標結果差于其他兩種特征單獨使用情況,藥物的蛋白質結構DBP特征和藥物在網絡中的位置信息SSE特征表現出相似的指標結果。指標結果波動情況也是PubChem在低維度時波動最大,SSE會略優于DBP特征。
同時,實驗發現使用線性分類器分類結果作為預測分數時計算AUC指標和AP指標的結果會更優。
表2是三種特征單獨使用,在最優參數設置下的各指標結果:
其中auc1表示使用預測結果作為預測分數時AUC指標的值,auc2則表示使用線性分類器分類分數作為預測分數時AUC指標的值。以此類推其他指標。
本文還考慮通過拼接這些特征得到更多的藥物信息進行DDIs預測實驗,這里使用最好預測效果的DBP和SSE進行拼接然后實驗,還考慮了將這三者進行拼接進行實驗,實驗結果如表3所示:
實驗指標和單獨采用SSE的情況沒有太高的優化,側面反映SSE應該是其中最有效果的特征信息。
表4是采用其他三個數據集得到的實驗結果和DB3的匯總,在這些實驗中,統一使用了SSE特征作為藥物節點的輸入特征表示。
6 結束語
本文使用圖符號網絡模型在公開藥物數據集上進行藥物相互關系預測實驗:通過對于藥物結點間正向邊和負向邊的不同處理聚合藥物圖中藥物鄰接點信息,獲得藥物的嵌入特征表達;通過拼接兩個藥物結點的特征表達的方法使用線性分類器進行分類預測。在已有公開數據集上的實驗結果表明,本文所述方法是有效的,亦是對現有方法的有益補充。
參考文獻:
[1] Shi J Y,Huang H,Li J X,et al.TMFUF:a triple matrix factorization-based unified framework for predicting comprehensive drug-drug interactions of new drugs[J].BMC Bioinformatics,2018,19(Suppl 14):411.
[2] 劉光徽,胡俊,於東軍.基于多視角特征組合與隨機森林的G蛋白 偶聯受體與藥物相互作用預測[J].南京理工大學學報(自然科學版),2016,40(1):1-9.
[3] 劉寧寧,琚生根,熊熙,等.基于膠囊網絡的藥物相互作用關系抽取方法[J].中文信息學報,2020,34(1):80-86,96.
[4] Feng Y H,Zhang S W,Shi J Y.DPDDI:a deep predictor for drug-drug interactions[J].BMC Bioinformatics,2020,21(1):419.
[5] Kipf T N, Welling M. Semi-supervised classification with graph convolutional networks[J]. arXiv preprint arXiv:1609.02907, 2016.
[6] Derr T,Ma Y,Tang J L.Signed graph convolutional networks[C]//2018 IEEE International Conference on Data Mining.November 17-20,2018,Singapore.IEEE,2018:929-934.
[7] Yu H,Mao K T,Shi J Y,et al.Predicting and understanding comprehensive drug-drug interactions via semi-nonnegative matrix factorization[J].BMC Systems Biology,2018,12(Suppl 1):14.
[8] Shi J Y,Mao K T,Yu H,et al.Detecting drug communities and predicting comprehensive drug-drug interactions via balance regularized semi-nonnegative matrix factorization[J].Journal of Cheminformatics,2019,11(1):28.
【通聯編輯:唐一東】
猜你喜歡
中國教育技術裝備(2016年19期)2016-12-27 19:23:52
中國遠程教育(2016年11期)2016-12-27 18:07:31
現代商貿工業(2016年25期)2016-12-26 09:58:02
江蘇教育·中學教學版(2016年11期)2016-12-21 11:45:08
江蘇教育·中學教學版(2016年11期)2016-12-21 11:36:29
現代情報(2016年10期)2016-12-15 11:50:53
考試周刊(2016年94期)2016-12-12 12:15:04
新教育時代·教師版(2016年23期)2016-12-06 06:02:38
法制與社會(2016年32期)2016-12-01 15:25:53
軟件導刊(2016年9期)2016-11-07 22:20:49
