基于加權基因共表達網絡分析篩選阿爾茨海默病的血液關鍵基因
2022-09-28 07:52:38魏冕張亞恒韓亞齊俊麗
實用心腦肺血管病雜志 2022年10期
魏冕,張亞恒,韓亞,齊俊麗
阿爾茨海默病(Alzheimer's disease,AD)是一種進展隱匿且病程不可逆的神經退行性疾病,至2019年全球AD患者人數已達5 000萬[1-2],且其死亡率呈逐年升高趨勢[3]。AD可能是由多種因素導致的復雜病理過程,且其早期診斷和治療研究進展緩慢。從基因層面分析,AD被認為是由大量基因在整個基因組[4]或轉錄組水平[5]上調控異常所致。因此,未來有望通過對異常表達的基因進行標記和干預來達到早期診斷和治療AD的目的。加權基因共表達網絡分析(weighted gene co-expression network analysis,WGCNA)已被證實可以有效檢測基因模塊與疾病特征之間的復雜關系[6]。WGCNA的獨特優勢是可以根據基因之間的權重相關系數將基因聚類到模型或網絡中,然后分析模塊與樣本特征(包括臨床特征、手術方法、治療方法等)之間的相關性。本研究通過對AD血液相關芯片數據進行分析并通過WGCNA篩選與AD相關的血液關鍵基因,旨在從基因層面為AD的診斷和治療提供新思路。
1 資料與方法
1.1 數據收集 2021年5—7月,利用美國國家生物技術信息中心基因表達綜合數據庫(https://www.ncbi.nlm.nih.gov/geo/)收集AD相關數據。所下載的數據集均以AD患者為實驗組,以年齡匹配的健康老年人為對照組,使用的是血液樣本。
1.2 基因注釋和差異表達基因(differentially expressed genes,DEG)的鑒定 將所有數據集的基因名稱進行重新注釋,刪除不能識別及重復識別的序列。在R 3.0.1軟件中,使用R-limma軟件包對兩組血液樣本中的mRNA進行差異表達分析。利用Benjamini-Hochberg方法進行多次測試校正。當mRNA的P<0.05、錯誤發現率(false discovery rate,FDR)<0.05和|log2 FC|≥1時被認為是DEG。
1.3 共表達網絡構建和模塊識別 使用WGCNA R包構建共表達網絡。首先,聚類評估樣本是否存在明顯異常值;其次,使用自動網絡構建功能構建共表達網絡。計算軟閾值β,根據β值獲得臨近矩陣和拓撲矩陣并計算基因之間的相異度。利用相異度對基因進行聚類,形成不同特征的模塊基因。根據性狀與模塊特征向量基因的相關性及P值來挖掘與性狀相關的模塊,選擇相關系數最高和P值最小的模塊。提取相應模塊的基因信息進一步分析。
1.4 功能富集分析 利用注釋、可視化和集成發現的數據庫(the database for annotation,visualization and integrated discovery,DAVID)對相關性最強的網絡模塊中的基因進行GO富集分析和KEGG通路富集分析,其中GO富集分析從生物學過程(biological process,BP)、細胞組成(cellular component,CC)和分子功能(molecular function,MF)3個方面進行。
1.5 關鍵基因鑒定 計算臨床相關性較強的幾個基因模塊的基因顯著性(gene significance,GS)(基因與性狀的相關性)和模塊身份(module membership,MM)(基因與模塊的相關性),將各模塊網絡中GS>0.9和MM>0.9的基因定義為核心基因。將核心基因上傳至STRING數據庫進行在線分析并建立蛋白-蛋白相互作用(protein protein interaction,PPI)網絡。在生物網絡中通過量化關鍵節點與非關鍵節點在拓撲性質方面的差異,并以量化值的大小作為判別關鍵節點與否的標準。使用Cytoscape的cytoHubba插件可計算網絡中各節點的最大團中心性(maximal clique centrality,MCC)、最大相鄰成分(maximum neighborhood component,MNC)、節點連接度(degree)、邊緣滲濾分量(edge percolated component,EPC)等拓撲特征。為了篩選出在PPI網絡中起關鍵作用的基因,并盡可能使結果穩健,利用該插件集成了多種網絡拓撲算法來識別網絡中的關鍵節點優點,將多種算法篩選出的前10個關鍵節點進行相交,最終獲得在多個算法中均處于關鍵節點的關鍵基因。
2 結果
2.1 DEG的鑒定 本研究數據集為GSE97760隊列的19個血液樣本,其中實驗組10個血液樣本、對照組9個血液樣本。使用FDR<0.05和|log2FC|≥1作為臨界點共鑒定出7 080個DEG,其中3 278個下調DEG和3 802個上調DEG,見圖1。
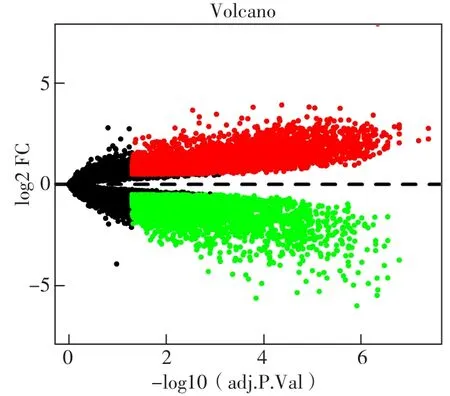
圖1 火山圖Figure 1 Volcano plot
2.2 共表達網絡構建和模塊識別 當β值為19時,基因間的連通性開始符合冪律分布(無尺度分布),因此可使用該閾值計算基因之間的相異度并構建WGCNA共表達網絡。將最小模塊大小設置為5,最終拆分出4個基因共表達模塊。結果顯示,黑色模塊 與AD呈正相關(r=0.89),綠色模塊與AD呈負相關(r=-0.90),見圖2。
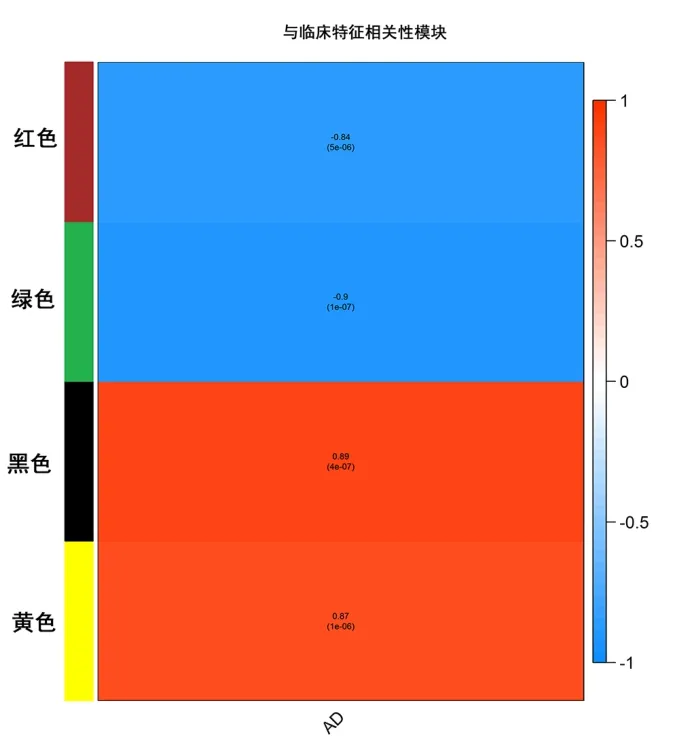
圖2 臨床特征相關性模塊圖Figure 2 Correlation module diagram of clinical features
2.3 關鍵模塊的功能富集分析 GO富集分析結果顯示,黑色模塊和綠色模塊基因的BP主要富集于轉錄、參與泛素依賴性蛋白質分解代謝過程的蛋白質泛素化,CC主要富集于核、核質,MF主要富集于蛋白質結合、DNA結合、泛素蛋白轉移酶活性。KEGG通路富集分析結果顯示,黑色模塊和綠色模塊基因主要調節途徑包括PI3K-Akt、MAPK信號通路。
2.4 關鍵基因鑒定 黑色模塊包含1 723個基因,綠色模塊包含2 321個基因。以GS>0.9和MM>0.9為臨界標準,在黑色模塊中鑒定出112個基因為核心基因,見圖3A;在綠色模塊中鑒定出42個基因為核心基因,見圖3B。將綠色模塊中的核心基因導入STRING數據庫后發現基因間交互關系稀少,基因間關系離散,故無法進一步鎖定關鍵基因。在黑色模塊中共得到4個關鍵基因(CUL5、RBM25、SRSF10、SRSF2),其中CUL5的MCC、MNC、degree、EPC均最大,見表1。

表1 黑色模塊中關鍵基因的拓撲特征Table 1 Topological characteristics of key genes in black module
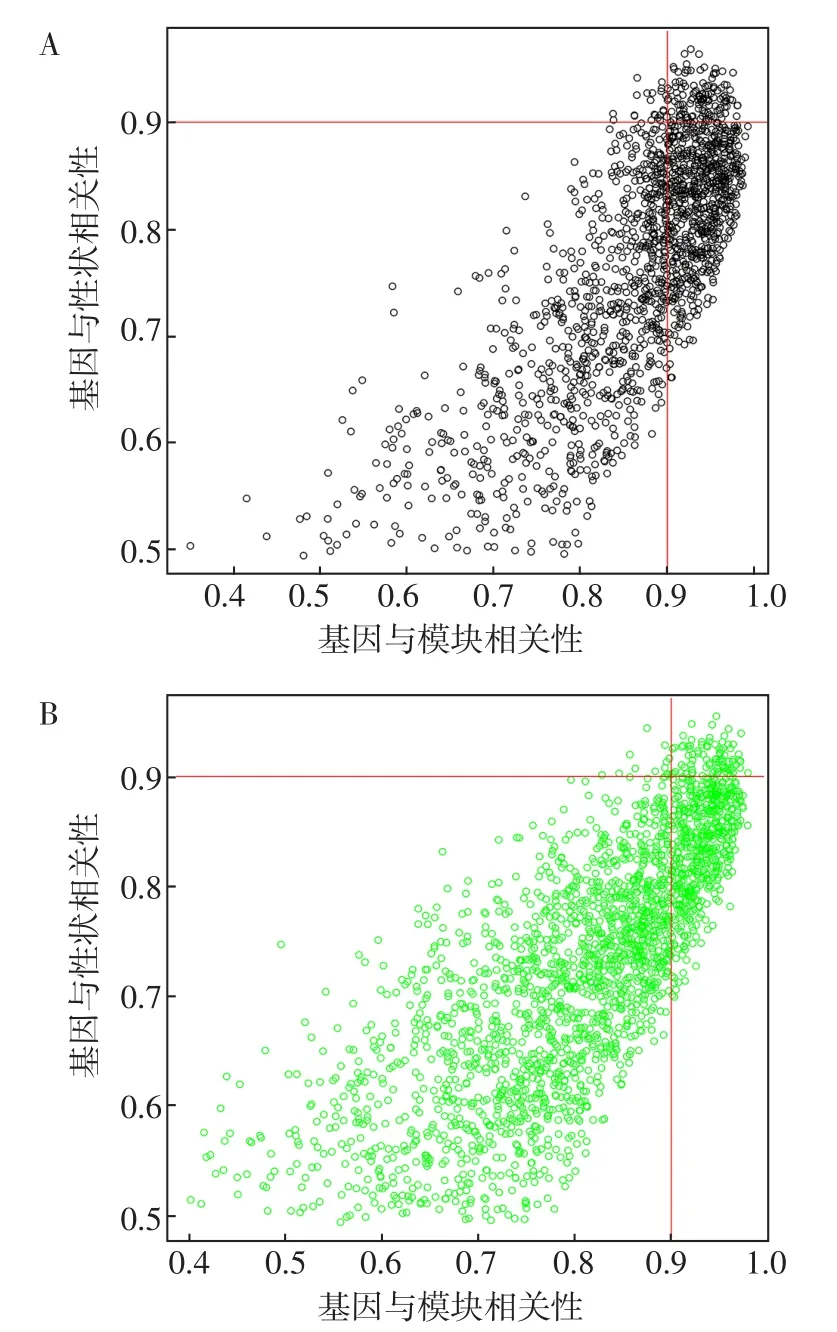
圖3 相關性最強的基因模塊的核心基因散點圖Figure 3 Scatter plot of the most correlated gene modules hub geens
3 討論
雖然正電子發射計算機斷層顯像及腰椎穿刺腦脊液檢查對AD具有較好的診斷效能[7],但昂貴的價格和有創操作等問題限制了其在AD早期診斷中的應用。本研究旨在鑒定AD患者血液中異常表達的關鍵基因,以從基因層面尋找新的生物標志物。
本研究通過構建共表達網絡從相關性最強的2個模塊中篩選出多個關鍵基因,其中在黑色模塊中通過多種算法篩選出的CUL5的中心特性最強,故認為其在模塊中的位置最為關鍵。既往研究發現,CUL5可以通過不同途徑參與AD的發生和發展,如CUL5過表達可減少腺苷酸環化酶和環磷酸腺苷的產生,通過絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)磷酸化的機制,參與與細胞增殖有關的各種蛋白質的降解過程,從而抑制細胞增殖[8-9];不僅如此,CUL5過表達還會導致DEPTOR水平明顯降低[10]。而高DEPTOR表達對于維持PI3K/Akt信號通路激活是必要的,此外其又是哺乳動物雷帕霉素靶蛋白復合體1(mammalian target of rapamycin complex 1,mTORC1)的抑制劑,可以抑制mTORC1的功能并誘導自噬[11]。這些途徑已被證實在AD的發生和發展中發揮著關鍵作用[12]。此外,黑色模塊中的SRSF2基因是一個重要的調節剪接tau蛋白的前mRNA,其剪接失調經常導致神經變性疾病[13-14]。而綠色模塊中的MIB1、USP9X已被證實可激活Wnt/β-catenin信號傳導通路[15-16],該信號通路的激活有助于AD的治療[17]。
近期一項基于基因表達綜合數據庫的網絡藥理學研究發現,藥物可以通過調控CUL5等核心基因來干預PI3K-Akt、MAPK、泛素介導的蛋白水解等信號通路,進而發揮治療AD的作用[18];此外,另一項網絡藥理學研究也發現,通過干預PI3K-Akt、MAPK信號通路可達到治療AD的效果[19]。這些途徑早已被多數研究證實在AD的發生和發展中具有核心作用[20-21]。筆者認為處于模塊最關鍵位置的CUL5可通過PI3K-Akt、MAPK信號通路在AD的發生和發展中發揮重要作用。
綜上所述,CUL5是AD的血液關鍵基因,其可調控PI3K-Akt、MAPK信號通路,并有望成為AD潛在的診斷和治療靶點。上述研究結果有助于揭示AD在基因層面的發生、發展機制。但本研究尚存在一定局限性:(1)本研究僅納入單個芯片數據,且數據中包含的病例較少,導致結果可能不穩定;(2)嚴格的重注釋方法可能導致部分錯配的探針序列不能被注釋,而丟失一些功能性mRNA;(3)部分關鍵基因與AD的關系尚不明確,需進一步驗證。
作者貢獻:魏冕、張亞恒進行文章的構思與設計,研究的實施與可行性分析;魏冕、韓亞、齊俊麗進行數據收集、整理、分析;魏冕、韓亞進行結果分析與解釋;魏冕負責撰寫、修訂論文;張亞恒負責文章的質量控制及審校,并對文章整體負責、監督管理。
本文無利益沖突。
猜你喜歡
中學生數理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
人大建設(2019年9期)2019-12-27 09:06:30
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
NBA特刊(2014年7期)2014-04-29 00:44:03
終身教育研究(2014年5期)2014-02-28 01:23:06
中國商人(2013年1期)2013-12-04 08:52:52
兒童時代(2009年5期)2009-05-21 05:31:26
