3DOM La0.4Ce0.6FeO3 可見光催化劑的制備及非均相光芬頓催化協同降解亞甲基藍
2022-10-19 03:49:46張琴琴李再興陳曉飛祁浩杰
石油化工 2022年9期
張琴琴,李再興,陳曉飛,李 超,祁浩杰,岳 欣
(1. 河北科技大學 環境科學與工程學院,河北 石家莊 050018;2. 河北省污染防治生物技術實驗室,河北 石家莊 050018;3. 天俱時工程科技集團有限公司,河北 石家莊 050011)
印染廢水具有排放量大、難降解、有機物含量高及堿性強等特點,導致它的處理難度極大[1-2]。亞甲基藍是一種偶氮染料,由于它的芳香結構不易被破壞,生化法和化學氧化等傳統方法很難將其降解[3-4]。高級氧化技術通過外界能量(如光能、電能等)和物質(如臭氧、雙氧水等)的持續輸入,經過一系列物理化學過程,產生羥基自由基(·OH)[5]。由于·OH 氧化電位高達2.8 V,可以氧化廢水中大多數有機物,因此,高級氧化技術具有廣闊的應用前景[6-7]。光芬頓技術和光催化技術通過吸收光輻射而產生氧化性極強的自由基實現污染物的高效降解,具有反應條件溫和、適用范圍廣、處理效率高及對污染物破壞徹底等優勢[8-10]。傳統均相光催化劑活性較高,但存在適應pH 范圍窄、難以回收和鐵泥產生量大等問題[11-13]。常用的光催化劑(如TiO2)需要紫外光激發,而紫外光只占自然光的約5%,限制了它在可見光下的高效運行[14]。鈣鈦礦材料具備適當的電子結構,可將帶隙能量轉移到可見光吸收,同時它的晶體結構允許晶格畸變,強烈影響光生電荷載流子的分離并避免復合過程,因此,鈣鈦礦材料是TiO2的優良替代材料[15-16]。鈣鈦礦型氧化物是一類具有與天然鈣鈦礦相同立方晶型結構的復合氧化物,化學通式為ABO3,A 位和B 位可被相同或不同價態離子取代,用A1?xA′xB1?yB′yO3+δ表示[17-18]。鈣鈦礦型氧化物因晶型結構穩定、催化活性高及晶格適應陽離子取代的靈活性,在非均相催化領域具有廣闊的應用前景[19]。通過傳統溶膠-凝膠法或共沉淀法制備的鈣鈦礦催化劑多以納米級顆粒存在,顆粒聚集程度高,比表面積小,不利于活性位點的暴露,進而限制了它在催化領域的應用[20-21]。三維有序大孔(3DOM)材料具有孔徑大、分布均勻及排列高度有序的孔道結構[22],豐富的孔道結構可以增加催化劑的比表面積,有利于反應物與活性位點的接觸,進而提高催化劑的活性[23-24]。因此,3DOM 材料在構建新型高效催化劑領域潛力巨大。
本工作以聚苯乙烯(PS)微球為模板,采用模板法聯合溶膠-凝膠法制備3DOM 材料La0.4Ce0.6FeO3(記作3DOM La0.4Ce0.6FeO3),并以亞甲基藍為降解對象,研究了它的催化性能、穩定性和催化機理。
1 實驗部分
1.1 主要原料
La(NO3)3·6H2O、Ce(NO3)3·6H2O、Fe(NO3)3·9H2O、亞甲基藍、異丙醇、對苯醌、草酸銨、L-組氨酸、FeSO4·7H2O、無水乙醇、檸檬酸:分析純,國藥集團化學試劑有限公司;PS 微球:直徑2 μm,天津市永大化學試劑有限公司;雙氧水:質量分數為50%:天津市永大化學試劑有限公司。
1.2 催化劑制備
PS 微球分散處理:將2.0 g PS 微球置于10 mL 無水乙醇中,超聲30 min。
3DOM La0.4Ce0.6FeO3前體的制備:將La(NO3)3·6H2O,Ce(NO3)3·6H2O,Fe(NO3)3·9H2O和檸檬酸按摩爾比為0.4∶0.6∶1.0∶2.0 溶于少量去離子水中,超聲30 min;加入分散好的PS 微球,于500 r/min 下磁力攪拌60 min,將混合液在磁力攪拌下,于80 ℃蒸發至凝膠態;將凝膠在空氣氛圍中于100 ℃下干燥12 h,得到3DOM La0.4Ce0.6FeO3前體。
3DOM La0.4Ce0.6FeO3的制備:將3DOM La0.4Ce0.6·FeO3前體在空氣氛圍下,以2 ℃/min 的速率升溫至200 ℃煅燒2 h,再以2 ℃/min 的速率升溫至400 ℃煅燒2 h,最后以4 ℃/min 的速率升溫至750℃煅燒5 h,得到3DOM La0.4Ce0.6FeO3。
采用溶膠-凝膠法[25]制備La1-xCexFeO3(x=0.2,0.4,0.6,0.8)和LaFeO3。
1.3 測試與表征
采用北京恒久實驗設備有限公司的HCT-2 型微機差熱天平進行熱重分析,空氣流量為60 mL/min,從室溫升至800 ℃,升溫速率為10 ℃/min;采用美國PE 公司SPECTRUM TWO 型傅里葉變換紅外光譜儀(波數為100 ~900 cm-1)和日本理學公司Ultima IV 型X 射線衍射儀(CuKα射線,衍射角為10°~80°)分析催化劑的物相和結構;采用德國卡爾蔡司公司ZEISS MERLIN Compact 型掃描電子顯微鏡觀察試樣的表面形貌,試樣進行噴金處理;采用美國Micromeritics 公司的ASAP2460 型比表面積及孔隙度分析儀測試催化劑的比表面積;采用美國Thermo Scientific 公司Thermo Scientific K-Alpha 型X 射線光電子能譜儀分析催化劑表面元素價態分布;采用日本Shimadzu 公司2550 型紫外-可見漫反射光譜儀測試催化劑的吸收光譜,波長200 ~800 nm。
1.4 亞甲基藍降解實驗與分析方法
亞甲基藍降解實驗在500 mL 自制反應器中進行。配制50 mg/L 的亞甲基藍溶液,用5%(w)HCl 及5%(w)NaOH 溶液調節體系的pH;投加催化劑,保持500 r/min 磁力攪拌,暗吸附60 min;加入雙氧水,打開光源開始反應,在特定時間取樣4 mL,用0.45 μm 濾膜過濾,考察出水中亞甲基藍的質量濃度、化學需氧量(COD)和總有機碳(TOC)。將過濾出的催化劑用去離子水反復清洗3 次后循環使用。
采用紫外可見分光光度法測定亞甲基藍質量濃度,在最大吸收波長665 nm 處,亞甲基藍質量濃度與吸光度的關系見式(1)。

式中,y為吸光度;x為亞甲基藍質量濃度,mg/L。
脫色率按式(2)計算。

式中,η為脫色率,%;ρ0為亞甲基藍初始質量濃度,mg/L;ρt為t時刻亞甲基藍質量濃度,mg/L。
采用北京連華永興科技發展有限公司2952400型快速測定儀測定COD,檢測方法為重鉻酸鉀 法[26]。采 用 日 本Shimadzu 公 司TOC-VCPH型總有機碳測定儀測定TOC。采用美國Thermo Scientific 公司ICAP RQ 型電感耦合等離子體質譜儀測定水中La,Ce,Fe 的質量濃度。
2 結果與討論
2.1 催化劑的表征結果
2.1.1 TG 分析
圖1 為3DOM La0.4Ce0.6FeO3前體的TG 曲線。由圖1 可知,3DOM La0.4Ce0.6FeO3前體失重過程分為7 個階段:第1 階段為25 ~150 ℃,質量損失率為2.86%,主要是水和無水乙醇的脫除;第2 階段為150 ~300 ℃,質量損失率為9.73%,主要是檸檬酸分解;第3 階段為300 ~350 ℃,質量損失率為3.06%,主要是剩余檸檬酸和部分PS 微球分解;第4 階段為350 ~500 ℃,質量損失率為30.59%,主要是PS 微球分解;第5 階段為500 ~550 ℃,質量損失率為3.18%,主要是剩余PS 微球分解和部分硝酸鹽分解;第6 階段為550 ~700 ℃,質量損失率為8.45%,主要是硝酸鹽分解;第7 階段為700 ℃以上,質量趨于穩定,表明試樣已經達到較為穩定的晶化程度,由此判斷鈣鈦礦結構在700 ℃以上生成。因此,將3DOM La0.4Ce0.6FeO3前體在200 ℃下煅燒2 h,400 ℃下煅燒2 h,750 ℃下煅燒5 h,可得到3DOM La0.4Ce0.6FeO3。

圖1 3DOM La0.4Ce0.6FeO3 前體的TG 曲線Fig.1 TG curve of the three-dimensional ordered macroporous(3DOM) La0.4Ce0.6FeO3 precursor.
2.1.2 物相及結構分析
圖2 為3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3的FTIR 譜圖。由圖2 可知,3DOM La0.4Ce0.6FeO3,La1-xCexFeO3與LaFeO3的 譜 線 均 在540 cm?1附 近出現Fe—O 鍵的非對稱伸縮振動吸收峰,代表典型的FeO6八面體簇,證明所制備催化劑均呈現鈣鈦礦結構[27]。

圖2 3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3 的FTIR 譜圖Fig.2 FTIR spectra of 3DOM La0.4Ce0.6FeO3,
圖3 為3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3的XRD 譜圖,由圖3A 可知,LaFeO3出現屬于鈣鈦礦結構的特征衍射峰(PDF#75-0541),未見明顯雜峰,由此可見LaFeO3為單一鈣鈦礦結構。La1-xCexFeO3與LaFeO3幾乎在相同位置出峰,表明La1-xCexFeO3已經形成鈣鈦礦結構。當x=0.8時,出現屬于CeO2的雜峰(PDF#75-0120),表明La0.2Ce0.8FeO3催化劑中形成CeO2雜質。由圖3B 可知,由于Ce3+,Ce4+的半徑略小于La3+,隨摻雜比例增大,衍射峰向大角度方向輕微偏移,表明Ce3+和Ce4+成功摻雜到鈣鈦礦結構中。3DOM La0.4Ce0.6FeO3與La0.4Ce0.6FeO3的出峰位置幾乎相同,表明3DOM La0.4Ce0.6FeO3同樣呈現單一鈣鈦礦結構。與LaFeO3相比,La1-xCexFeO3峰形變寬,強度下降,表明摻雜Ce 離子使材料的結晶度降低,對鈣鈦礦結構有一定程度的破壞作用[28]。在La1-xCexFeO3中,La0.4Ce0.6FeO3的衍射峰最強最尖銳,表明當x=0.6 時,催化劑結晶度最高。與La0.4Ce0.6FeO3相比,3DOM La0.4Ce0.6FeO3衍射峰強度和尖銳程度明顯更高,表明3DOM La0.4Ce0.6FeO3具有比La0.4Ce0.6FeO3更高的結晶度和晶體穩定性,這可能是因為3DOM 結構使晶體具有更大的生長空間[29]。
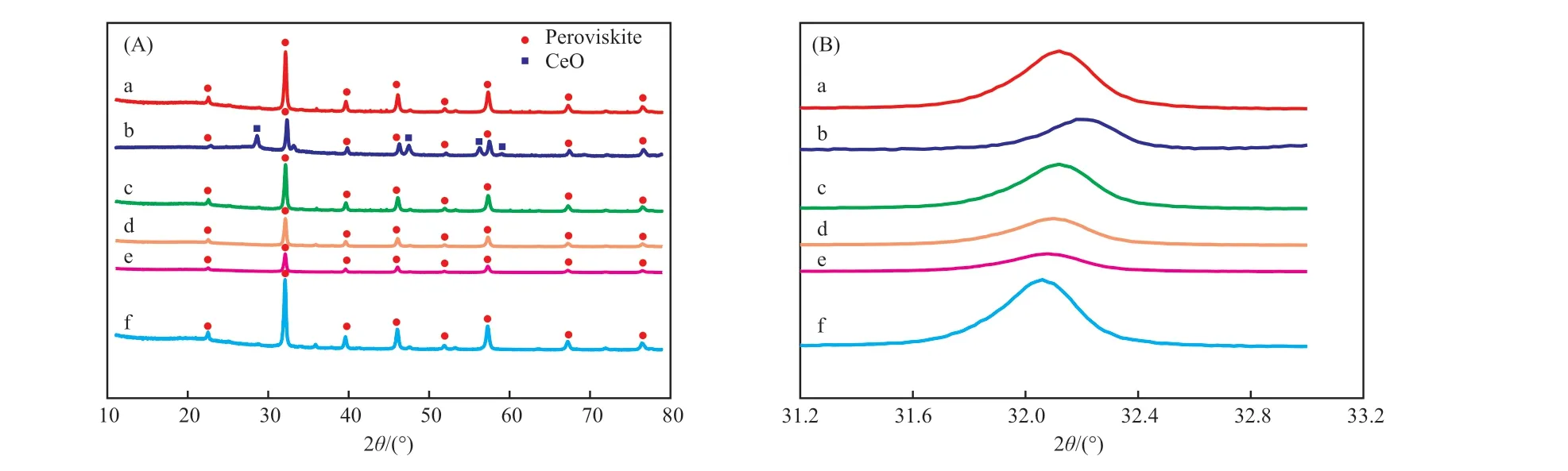
圖3 催化劑的廣角XRD 譜圖(A)與小角XRD 譜圖(B)Fig.3 WAXD patterns of the catalysts(A) and the SAXD patterns of the catalysts(B).
由謝樂公式(式(3))計算催化劑的鈣鈦礦晶粒尺寸:
D=Kλ/(βcosθ) (3)
式 中,D為 晶 粒 粒 徑,nm;λ為 衍 射 波 長,λ=0.154 1 nm;β為半峰寬,°;θ為晶格衍射角,°;K為常數0.890 0。
表1 為催化劑晶粒粒徑。由表1 可知,La1-xCexFeO3晶粒粒徑小于LaFeO3,這是因為整個體系為保持整體的電價平衡,會有部分晶格氧空位出現,為保持鈣鈦礦結構,La1-xCexFeO3晶胞出現一定收縮,因而晶胞體積有所減小[30]。3DOM La0.4Ce0.6FeO3晶粒粒徑大于La0.4Ce0.6FeO3,進一步表明摻雜對鈣鈦礦晶體結構有一定程度的破壞作用,以及3DOM 結構有利于晶體結構的穩定。
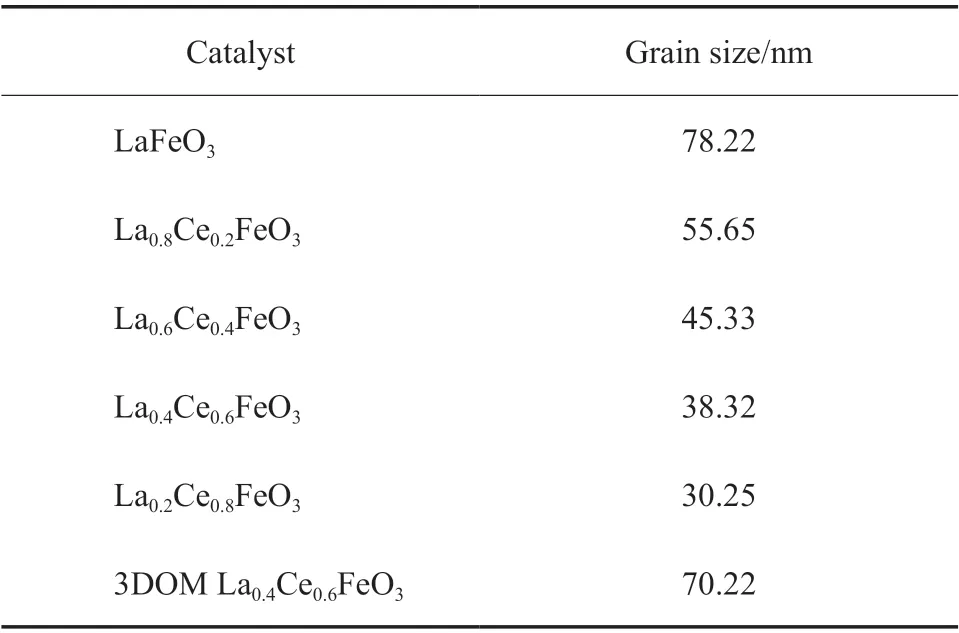
表1 催化劑的晶粒粒徑Table 1 Grain size of the catalysts
2.1.3 表面形貌分析
圖4 為PS 微球,3DOM La0.4Ce0.6FeO3,La0.4Ce0.6FeO3的SEM 照 片。 由 圖4 可 知,PS 微 球 形 貌 規整,粒徑約為2 μm;采用溶膠-凝膠法制備的La0.4Ce0.6FeO3晶體顆粒高度團聚,形成表面光滑的塊狀結構,這種結構限制了催化劑的比表面積;3DOM La0.4Ce0.6FeO3呈現3DOM 結構,孔徑約為2 μm,與PS 微球粒徑相當;與La0.4Ce0.6FeO3相比,3DOM 結構使催化劑比表面積得到顯著提高。綜上所述,3DOM 結構有利于活性位點的暴露,進而提升催化劑活性[31]。

圖4 PS 微球(a),La0.4Ce0.6FeO3(b),3DOM La0.4Ce0.6FeO3(c)的SEM 照片Fig.4 SEM images of polystyrene microsphere(a),La0.4Ce0.6FeO3(b) and 3DOM La0.4Ce0.6FeO3(c).
2.1.4 比表面積分析
3DOM La0.4Ce0.6FeO3的比表面積(70.890 m2/g)是La0.4Ce0.6FeO3比表面積(5.231 m2/g)的13.55 倍,表明3DOM 結構顯著提升了催化劑的比表面積,這與SEM 表征結果一致。
2.1.5 活性組分元素價態分析
圖5 為3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3的XPS 譜圖。對Fe 2p3/2軌道和Ce 3d軌道進行分峰擬合,結果見表2。由圖5 可知,對Fe 2p3/2軌道進行分峰擬合,LaFeO3中Fe 以Fe3+形式存在,而La1-xCexFeO3中存在Fe2+。大量研究結果表明,Fe2+在光芬頓中的催化活性高于Fe3+[32]。由圖5 還可知,La1-xCexFeO3中Ce3+與Ce4+是共同存在的,原子分數分別為15%和85%左右。在La1-xCexFeO3中同時存在Fe2+,Fe3+,Ce3+,Ce4+,根據電中性原則,La1-xCexFeO3中存在氧空位,而氧空位對光芬頓過程具有促進作用[33-34]。基于以上兩點,Ce 的摻雜有利于提高鈣鈦礦催化劑的活性。

圖5 催化劑的XPS 譜圖Fig.5 XPS spectra of the catalysts.
由 表2 可 知, 當x=0.6 時,La0.4Ce0.6FeO3中Fe2+含量為100.0%,催化劑表面氧空位也最為豐富,所以此時摻雜催化劑具有最高活性。3DOM La0.4Ce0.6FeO3中Fe 同樣以Fe2+形式存在,氧空位含量也與La0.4Ce0.6FeO3相當,表明3DOM 結構不會對鈣鈦礦材料的活性產生不利影響。

表2 催化劑表面活性組分的價態Table 2 Valence of active components on catalyst surface
2.1.6 吸光性能分析
圖6 為3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3的吸收光譜。由圖6 可知,采用溶膠-凝膠法制備的LaFeO3在波長為200 ~800 nm 時均有明顯的吸收帶,La1-xCexFeO3具有比LaFeO3更高的光響應強度和較寬的光響應波長范圍,表明摻雜Ce 有利于改善LaFeO3的光學性能。當x=0.6 時,La0.4Ce0.6FeO3吸光強度較強,說明此時對光的吸收能力好。3DOM La0.4Ce0.6FeO3具有比La0.4Ce0.6FeO3更高的吸光能力,表明3DOM 結構有利于增加催化劑的比表面積,增強光透過性,提高光吸收能力,進而提高光催化活性[35]。3DOM La0.4Ce0.6FeO3在400 ~600 nm 吸光強度最大,該范圍在可見光區,實驗選用420 nm 氙燈。

圖6 3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3 的吸收光譜Fig.6 3DOM La0.4Ce0.6FeO3,La1-xCexFeO3 and LaFeO3 absorption spectra.
2.2 3DOM La0.4Ce0.6FeO3 的性能
2.2.1 吸附性能
當反應溫度為25 ℃、亞甲基藍初始質量濃度為50 mg/L、3DOM La0.4Ce0.6FeO3投加量為500 mg/L時,在黑暗條件下,3DOM La0.4Ce0.6FeO3的脫色率、COD 去除率和TOC 去除率隨吸附時間的變化見圖7。由圖7 可知,在不同pH 下均去除了一定的亞甲基藍,表明在測試范圍內(pH=2 ~10),3DOM La0.4Ce0.6FeO3有一定的吸附性能。隨pH 降低,亞甲基藍去除效率逐漸上升,表明3DOM La0.4Ce0.6FeO3在較低pH 條件下具有更強的吸附性能,這可能是因為亞甲基藍在水中解離帶負電,當溶液呈酸性時,催化劑獲得氫離子的能力增強而帶正電,導致靜電作用增強,從而吸附能力增強[36]。在測試范圍內,脫色率、COD 去除率和TOC 去除率均在40 ~60 min 時達到最高后趨于平緩,表明不同pH 條件下均在40 ~60 min 達到吸附平衡,所以后續實驗均選擇暗吸附時間為60 min。

圖7 脫色率、COD 去除率和TOC 去除率隨吸附時間的變化Fig.7 The variation of decolorization rate,COD removal rate and total organic carbon(TOC) removal rate with adsorption time.
2.2.2 初始pH 的影響
在420 nm 可見光、反應溫度為25 ℃、亞甲基藍初始質量濃度為50 mg/L、雙氧水投加量為0.5 mL/L、3DOM La0.4Ce0.6FeO3投加量為500 mg/L、暗吸附時間為60 min 的條件下,初始pH 對脫色率、COD 去除率和TOC 去除率的影響見圖8。由圖8 可知,FeSO4·7H2O 作為均相催化劑,當初始pH 在2 ~4 時,脫色率、COD 去除率和TOC 去除率分別在90%,85%,80%以上,表明均相催化劑在低pH 范圍內具有較高的活性;當初始pH 為4 ~10 時,亞甲基藍降解效率明顯下降;當pH 為10 時,脫色率、COD 去除率和TOC 去除率分別低至20.6%,20.0%,13.0%,表明均相催化劑適應pH 范 圍 窄。3DOM La0.4Ce0.6FeO3,La1-xCexFeO3,LaFeO3作為非均相催化劑,在初始pH 為2 ~10時,對亞甲基藍的降解效率均高于FeSO4·7H2O,表明鈣鈦礦催化劑活性較高且pH 適用范圍較廣。在La1-xCexFeO3中,當x=0.6 時,La0.4Ce0.6FeO3呈現最高的亞甲基藍降解效率,表明在此摻雜比例下鈣鈦礦催化活性最高。3DOM La0.4Ce0.6FeO3比La0.4Ce0.6FeO3的催化活性更高,表明3DOM 結構使鈣鈦礦催化劑的活性得到進一步提升,這與催化劑表征結果一致。

圖8 初始pH 對脫色率、COD 去除率和TOC 去除率的影響Fig.8 The influence of initial pH on decolorization rate,COD removal rate and TOC removal rate.
采用ICP-MS 檢測出水Fe 質量濃度。當pH為2,3,4,5,6,7,8,9,10 時,使用3DOM La0.4Ce0.6FeO3催化劑,Fe 浸出質量濃度分別為0.45,0.32,0.25,0.20,0.19,0.06,0.02,0,0 mg/L,均低于0.50 mg/L,明顯低于使用均相催化劑時的Fe 質量濃度(112 mg/L),表明3DOM La0.4Ce0.6FeO3作為非均相光催化劑可以減少鐵泥的產生,避免二次污染,降低處理成本。
2.2.3 雙氧水投加量的影響
雙氧水利用率是非均相光芬頓反應中一項重要的評估參數[37]。在420 nm 可見光、反應溫度為25℃、亞甲基藍初始質量濃度為50 mg/L,初始pH 為3、3DOM La0.4Ce0.6FeO3投加量為500 mg/L、暗吸附時間為60 min 的條件下,雙氧水投加量對脫色率、COD 去除率和TOC 去除率的影響見圖9。由圖9可知,隨雙氧水投加量減少,亞甲基藍的降解效率均呈下降趨勢,這可能是因為雙氧水是自由基的主要來源,雙氧水投加量減少,自由基產生量下降,進而限制了亞甲基藍的降解[38]。當雙氧水投加量為0.1 mL/L 時,脫色率、COD 去除率和TOC 去除率分別達到90.6%,60.5%,51.0%,顯著高于其他催化劑,這表明3DOM La0.4Ce0.6FeO3具有最高的雙氧水利用率。這可能是因為3DOM 結構使催化劑比表面積增大,豐富的孔道和較大的比表面積增強了光的捕獲與傳質,進而提升了催化劑的活性[39]。

圖9 雙氧水投加量對脫色率、COD 去除率和TOC 去除率的影響Fig.9 The influence of hydrogen peroxide dosage on decolorization rate,COD removal rate and TOC removal rate.
2.3 3DOM La0.4Ce0.6FeO3 的穩定性
在420 nm 可見光、反應溫度25 ℃、亞甲基藍初始質量濃度50 mg/L、初始pH 為3、雙氧水投加量0.5 mL/L、3DOM La0.4Ce0.6FeO3投加量500 mg/L、暗吸附時間為60 min 的條件下進行重復性實驗,3DOM La0.4Ce0.6FeO3光芬頓協同光催化降解的穩定性見圖10。由圖10 可知,3DOM La0.4Ce0.6FeO3經過5 次重復使用之后,脫色率、COD 去除率和TOC 去除率分別維持在98.7%,90.0%,80.5%。表明3DOM La0.4Ce0.6FeO3穩定性較高。

圖10 3DOM La0.4Ce0.6FeO3 光芬頓協同光催化降解的穩定性Fig.10 The stability of 3DOM La0.4Ce0.6FeO3 photocatalysis-Fenton synergistic degradation.
催化劑使用前與重復使用5 次后的SEM 照片及XRD 譜圖見圖11。由圖11 可知,3DOM La0.4Ce0.6FeO3催化劑重復使用5 次后,孔結構雖然發生了一定程度的坍塌,但仍呈現3DOM 結構,表明3DOM La0.4Ce0.6FeO3結構穩定性較強;重復使用5 次后,催化劑仍呈現鈣鈦礦的特征峰,表明鈣鈦礦的晶體結構沒有破壞,同時鈣鈦礦特征峰的強度和尖銳程度沒有明顯變化,表明材料結晶度在反應過程中也沒有明顯變化。

圖11 催化劑使用前(a)與重復使用5 次后(b)的SEM 照片及XRD 譜圖(c)Fig.11 SEM images of catalyst before used(a) and catalyst after reused 5 times(b),and XRD spectra of the catalyst(c).
利用ICP-MS 檢測出水中La,Ce,Fe 的含量,計算浸出量,結果見表3。由表3 可知,在重復實驗過程中,La,Ce,Fe 的浸出量均在0.042 5%(w)以下,表明活性組分不易流失,催化劑穩定性較強。

表3 La,Ce 和Fe 的浸出量Table 3 Leaching of La,Ce and Fe
2.4 催化反應機理分析
2.4.1 催化反應機制
為研究亞甲基藍催化降解機制,利用猝滅實驗考察自由基的作用,選用異丙醇、對苯醌、草酸銨和L-組氨酸分別作為·OH,·O2-,h+,1O2的猝滅劑。猝滅劑對脫色率、COD 去除率和TOC 去除率的影響見圖12。由圖12 可知,h+和1O2被清除后,脫色率、COD 去除率和TOC 去除率沒有明顯變化,表明h+和1O2對亞甲基藍的降解作用不明顯;·OH或·O2-被清除后,脫色率、COD 去除率和TOC去除率均下降,表明·OH 和·O2-在亞甲基藍降解過程中起重要作用;·OH 和·O2-同時被清除后,脫色率、COD 去除率和TOC 去除率與只加催化劑相當,表明亞甲基藍的降解是在·OH 和·O2-共同作用下完成的。

圖12 猝滅劑對脫色率、COD 去除率和TOC 去除率的影響Fig.12 Influence of quenching agent on decolorization rate,COD removal rate and TOC removal rate.
為進一步證明催化體系中·OH 和·O2-的存在,采用電子自旋共振(ESR)技術分別對投加雙氧水前10 min 和后10 min 體系中的·OH 和·O2-進行測定,·OH 和·O2-的產生情況見圖13。由圖13 可知,在無雙氧水條件下,出現屬于DMPO-·OH(DMPO 為5,5-二甲基-1-吡咯啉-N-氧化物)加合物信號1∶2∶2∶1 四重峰和BMPO-·O2-(BMPO 為5-叔丁氧羰基-5-甲基-1-吡咯啉-N-氧化物)加合物信號1∶1∶1∶1 特征峰,加入雙氧水10 min 后,產生更強的DMPO-·OH加合物信號1∶2∶2∶1 四重峰和BMPO-·O2-加合物信號1∶1∶1∶1 特征峰,兩種特征峰分別代表·OH 和·O2-的存在。以上結果進一步證明亞甲基藍的降解是在·OH 和·O2-的共同作用下完成的。

圖13 ·OH(a)和·O2-(b)的產生情況Fig.13 Production of ·OH (a) and·O2- (b).
ESR 測試結果顯示,在加入雙氧水之前已經存在·OH 和·O2-的特征峰,表明實驗體系中存在光催化反應。為證實光催化反應的存在,考察了光催化、光芬頓和光芬頓協同光催化三種體系中脫色率、COD 去除率和TOC 去除率隨反應時間的變化,結果見圖14。
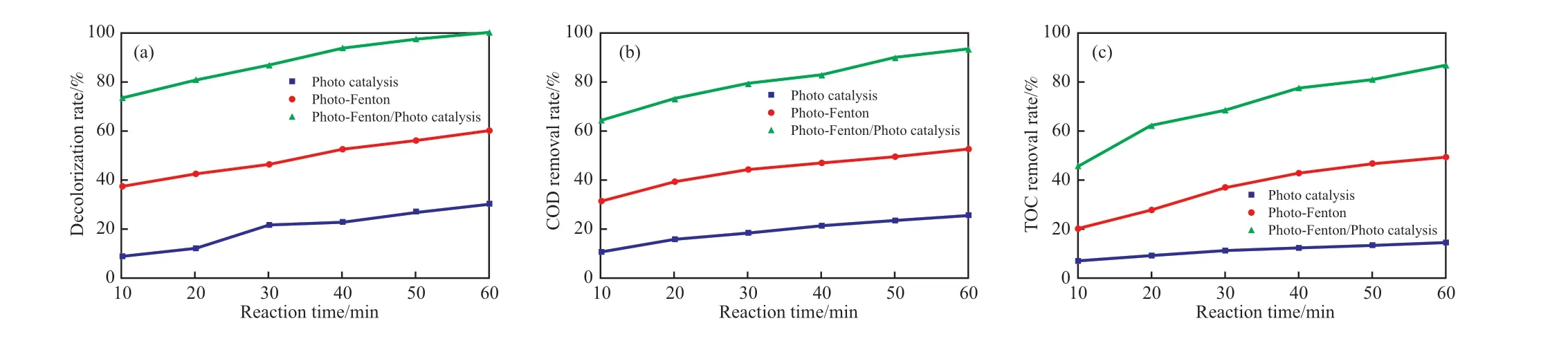
圖14 光催化、光芬頓和光芬頓-光催化三種體系中脫色率、COD 去除率和TOC 去除率隨反應時間的變化Fig.14 The variation of decolorization rate,COD removal rate and TOC removal rate with reaction time in photocatalytic,photo-Fenton and photo-Fenton/photocatalytic systems.
由圖14 可知,光芬頓體系和光催化體系均對亞甲基藍有一定去除效果,光芬頓協同光催化體系的降解效率高于光催化體系與光芬頓體系之和,表明在反應體系中存在光芬頓和光催化的協同作用。
2.4.2 自由基產生機理


圖15 自由基產生機理Fig.15 Mechanism of free radical production.
2.4.3 催化反應動力學
在420 nm 可見光、反應溫度25 ℃、亞甲基藍初始質量濃度50 mg/L、初始pH 為3、雙氧水投加量0.5 mL/L、3DOM La0.4Ce0.6FeO3投加量500 mg/L條件下,進行亞甲基藍降解實驗。在亞甲基藍完全脫色之前(反應時間小于30 min),將實驗數據用最小二乘法進行擬合。一級反應動力學曲線見圖16。由圖16 可知,催化反應動力學方程為ln(ρ0/ρt)=0.027 35t-0.069 63,相關系數為0.946 68,表明亞甲基藍的降解近似符合一級動力學反應。

圖16 一級反應動力學曲線Fig.16 First order reaction kinetic curve.
3 結論
1)采用模板法聯合溶膠-凝膠法成功制備了3DOM La0.4Ce0.6FeO3,實現了對亞甲基藍的高效降解。
2)3DOM結構增大了La0.4Ce0.6FeO3的比表面積,豐富的孔道結構和比表面積提高了La0.4Ce0.6FeO3對光的吸收能力和傳遞速率,從而提高了鈣鈦礦催化劑的活性。
3)3DOM La0.4Ce0.6FeO3具有比均相光催化劑更廣的pH 適用范圍、更少的鐵泥產量和更高的雙氧水利用率。
4)重復使用5 次之后,3DOM La0.4Ce0.6FeO3對亞甲基藍的降解效率沒有明顯下降,同時催化劑的3DOM 結構和鈣鈦礦晶形結構沒有明顯改變,催化劑穩定性較強。
5)亞甲基藍的降解是在·OH 和·O2-共同作用下完成的;在可見光照射下,同時存在光催化和光芬頓兩種反應,產生大量·OH 和·O2-實現了亞甲基藍的降解,Ce4+和Ce3+的循環反應提升了電子轉移效率,促進了自由基的產生;實驗數據擬合結果表明,亞甲基藍的降解近似符合一級動力學。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
