P摻雜BiOBr的制備及其光催化固氮性能研究
2022-12-19 12:48:44葛建華丁修龍
功能材料 2022年11期
葛建華,李 佳,丁修龍,衛 洲,張 萬
(1. 安徽理工大學 地球與環境學院,安徽 淮南 232001;2. 合肥綜合性國家科學中心能源研究院(安徽省能源實驗室), 合肥 230031)
0 引 言
近年來,能源短缺和環境破壞的嚴重挑戰制約了社會的可持續發展[1- 2]。眾所周知,傳統的哈伯-博世固氮過程需要消耗大量的電力和氫氣,同時也會排放大量的溫室氣體[3]。為了節約能源和保護環境,開發綠色人工固氮技術引起了研究者的關注[4-5]。
BiOBr具有層狀結構、帶隙(~2.9 eV)和可見光響應性能好[6],引起了廣泛的關注。然BiOBr存在太陽光利用率低、量子效率以及光生電子還原能力低等缺點[7]。為解決上述問題,科研工作者對其進行改性,如構造異質結[8],晶面調控[9],離子摻雜等[10]。如Maisang等[11]通過微波輔助水熱法在含聚乙烯吡咯烷酮(PVP)溶液中制備出BiOBr/BiOCl花狀復合材料,表現出優異的光催化性能。Sin等[12]采用水熱法將Nd成功摻雜到BiOBr中,大大改善光生載流子的分離效率。同時,也表現出優異的穩定性。此外,非金屬離子摻雜對光催化劑的催化效果有顯著的提升作用[13-18]。Chen等[19]將次亞磷酸鈉作為P源,成功將P元素摻雜到TiO2,顯著增強了光催化活性。
綜上所述。在本實驗中,采用溶劑熱法制備出P摻雜BiOBr。實驗表明,P摻雜促進光生電子-空穴的分離效率,進而提高其光催化固氮活性。循環實驗,說明P摻雜BiOBr的穩定性較好。
1 實 驗
1.1 實驗試劑
實驗所用化學試劑:溴化鉀(KB ,≥99.0%,分析純)、五水硝酸鉍(Bi(NO3)3·5H2O,≥99.0%,分析純)、乙醇(C2H6O, ≥99.7%,分析純)、乙二醇((CH2OH)2, ≥98%,分析純)、丙三醇(C3H8O3,≥99%,分析純)、1, 3-丁二醇(C4H10O2,≥99%,分析純)、四水合酒石酸鉀鈉(C4H4O6KNa·4H2O,≥99%,分析純)以及納氏試劑均采購于阿拉丁生化科技股份有限公司;一水次亞磷酸鈉(NaH2PO2·H2O,≥98%,分析純)采購于國藥集團化學試劑有限公司。
1.2 實驗儀器
X射線衍射儀(XRD,5°/min, 10°~80° in 2θ, Smartlab SE, Japan),掃描電子顯微鏡(SEM, FlexSEM 1000,Hitachi,日本),X射線光電子能譜儀(XPS,ESCALAB 250Xi,美國),比表面積和孔徑分布由Brunauer-Emmett-Teller(BET,ASAP2020,美國)、Barrett-Joyner-Halenda法計算獲得。此外,紫外-可見分光譜儀(LAMBDA 950,PerkinElmer,美國)和電化學阻抗譜法(EIS,CHI760E,上海成華科技有限公司)。
1.3 純BiOBr的制備
分別稱取1 mmol的Bi(NO3)3·5H2O和KBr置入于20mL乙二醇中攪拌溶解,待兩者均完全溶解后,再混合攪磁力拌均勻,放入反應釜中,在160℃下反應12 h,冷卻后離心,用去離子水及無水乙醇洗滌,烘干備用。
1.4 P摻雜改性BiOBr催化劑制備
將1 mmol的Bi(NO3)3·5H2O超聲溶于20 mL乙二醇。另外,分別稱取1 mmol的KBr和一定量的NaH2PO2加入20 mL乙二醇中,攪拌至完全溶解。3種溶液完全溶解后混合均勻,放入100 mL反應釜中,在160℃下反應12 h,冷卻后離心,用去離子水及無水乙醇洗滌后,烘干備用。其中,以Bi∶P的摩爾比來確定NaH2PO2的投加量,得到的樣品命名為P-xBiOBr。
1.5 光催化固氮實驗
本實驗采用300 W的氙燈光源模擬太陽光照,在室溫和常壓下,向250 mL自制的石英反應器中加入~0.05 g光催化劑、一定量捕獲劑和200 mL去離子水。同時,為保持整個反應過程中溶液和催化劑均勻混合,使用磁力攪拌器進行攪拌。采用空氣泵向反應器中通入空氣為反應體系提供氮源。在氙燈光源照射下反應,定時取10 mL樣品,離心后,取上清液待測。其中,分別依據納氏試劑分光光度法(HJ 535-2009)、亞硝酸鹽氮分光光度法(HJ GB 7493-87)和硝酸鹽氮紫外分光光度法(HJ /346-2007)檢測樣品中的氨氮、亞硝酸鹽氮和硝酸鹽氮含量。
2 催化劑表征分析
2.1 XRD分析
圖1為不同P摻雜量催化劑的XRD譜圖。催化劑突出特征峰與標準峰(JCPDS No.73-2061)相吻合,且無其他明顯雜峰,這說明P元素摻雜的BiOBr與原始BiOBr具有基本相同的晶體結構[20]。在2θ為25.25°、32.22°和33.13°對應的晶面分別是(101)、(102)和(110)。對比BiOBr,P元素摻雜的催化劑晶面的衍射峰顯著降低,說明P元素抑制了BiOBr向(102)晶面和(110)晶面的生長取向, 所以晶體的結晶度會隨著峰的強度減弱而下降。此外,與BiOBr材料相比,P摻雜BiOBr存在不同程度的峰寬化現象,可推斷P摻雜制備的BiOBr材料的粒徑要小。
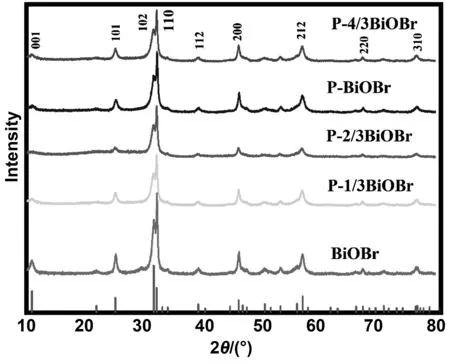
圖1 催化劑的XRD圖譜
2.2 SEM分析
如圖2所示BiOBr和P-BiOBr都是花瓣狀的球形體,構成花球的不規則納米片的厚度大約為10 nm。P-BiOBr的形狀和大小沒有發生很大改變,但其花瓣片之間可以明顯觀察到其它小顆粒附著在上面,層狀結構的尺寸在減少。這也間接說明P-BiOBr的XRD圖譜中特征衍射峰強度下降的原因,P-BiOBr的團聚度比原始BiOBr低,該結構能夠有效提高了光催化劑的固氮效果。
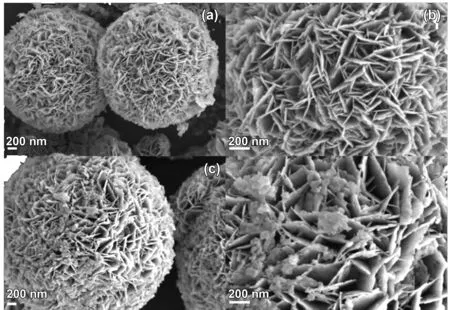
圖2 BiOBr(a),(b)和P-BiOBr(c),(d)的SEM圖
2.3 XPS分析

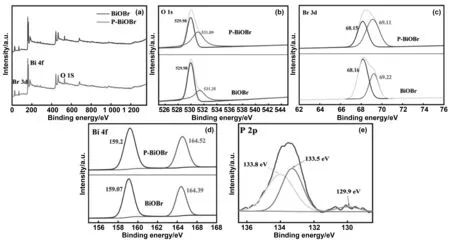
圖3 催化劑的XPS圖譜:(a)全譜;(b) O 1s;(c) Br 3d;(d)Bi 4f;(e)P 2p)
2.4 UV-Vis分析
為探究光催化劑的光學吸收性能,對催化劑進行了UV-Vis DRS測試。如圖4a所示,BiOBr和P-BiOBr均有明顯的吸收邊緣,BiOBr吸收波長大約為430 nm,P-BiOBr吸收波長大約為455 nm。如圖4b所示,BiOBr的帶隙能約為2.76 eV,P-BiOBr帶隙能約為2.62 eV,比原始催化劑的帶隙能小0.14 eV,有更好的可見光響應度,固氮效果也明顯更優。

圖4 催化劑的UV-Vis DRS譜圖
2.5 比表面積和孔徑分析
圖5可以看出原始BiOBr和P-BiOBr復合材料的等溫線(IV型)和滯回線(H3型)都相同,這表明兩種樣品均具有介孔結構。并且P-BiOBr的孔徑多數都為5.152nm,相比于BiOBr要小很多,但是BiOBr(0.0837 cm3/g)和P-BiOBr(0.0817 cm3/g)的孔容接近。用BET法計算的P-BiOBr的比表面積為20.779 m2/g,比BiOBr(15.383 m2/g) 的比表面積增大,這可能是P-BiOBr表面依附了很多小顆粒,提供了更多的反應活性位點,進而有利于光催固氮化反應的進行。

表1 BiOBr和P-BiOBr的孔徑,孔容及比表面積

圖5 催化劑的氮氣吸附-解吸等溫線及相應孔徑分布曲線
2.6 光電流電化學性能分析
圖6為催化劑的瞬態光電流響應曲線和EIS奈奎斯特曲線。兩種催化劑在光照條件下都產生瞬態光電流響應,在光照后一段時間內P-BiOBr光電流強度有所降低,這說明了部分載流子發生了復合。從圖6a中可以看出,P-BiOBr光電流強度大于BiOBr的光電流強度,說明P摻雜增強了BiOBr中e-/h+對的分離能力。由圖6b中看到P-BiOBr的EIS奈奎斯特曲線的圓弧半徑更大,這表明P摻雜沒有降低BiOBr電荷轉移的電阻,這可能是由于其表面附著的小顆粒所致,降低催化劑的電子傳導性[28]。但綜合兩個因素來看,P摻雜依然提高了催化劑的固氮效果。
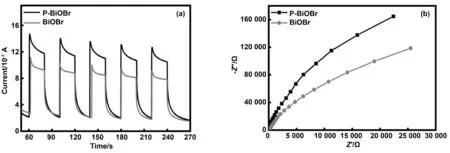
圖6 催化劑的瞬態光電流響應曲線和EIS奈奎斯特曲線
3 光催化固氮性能的研究
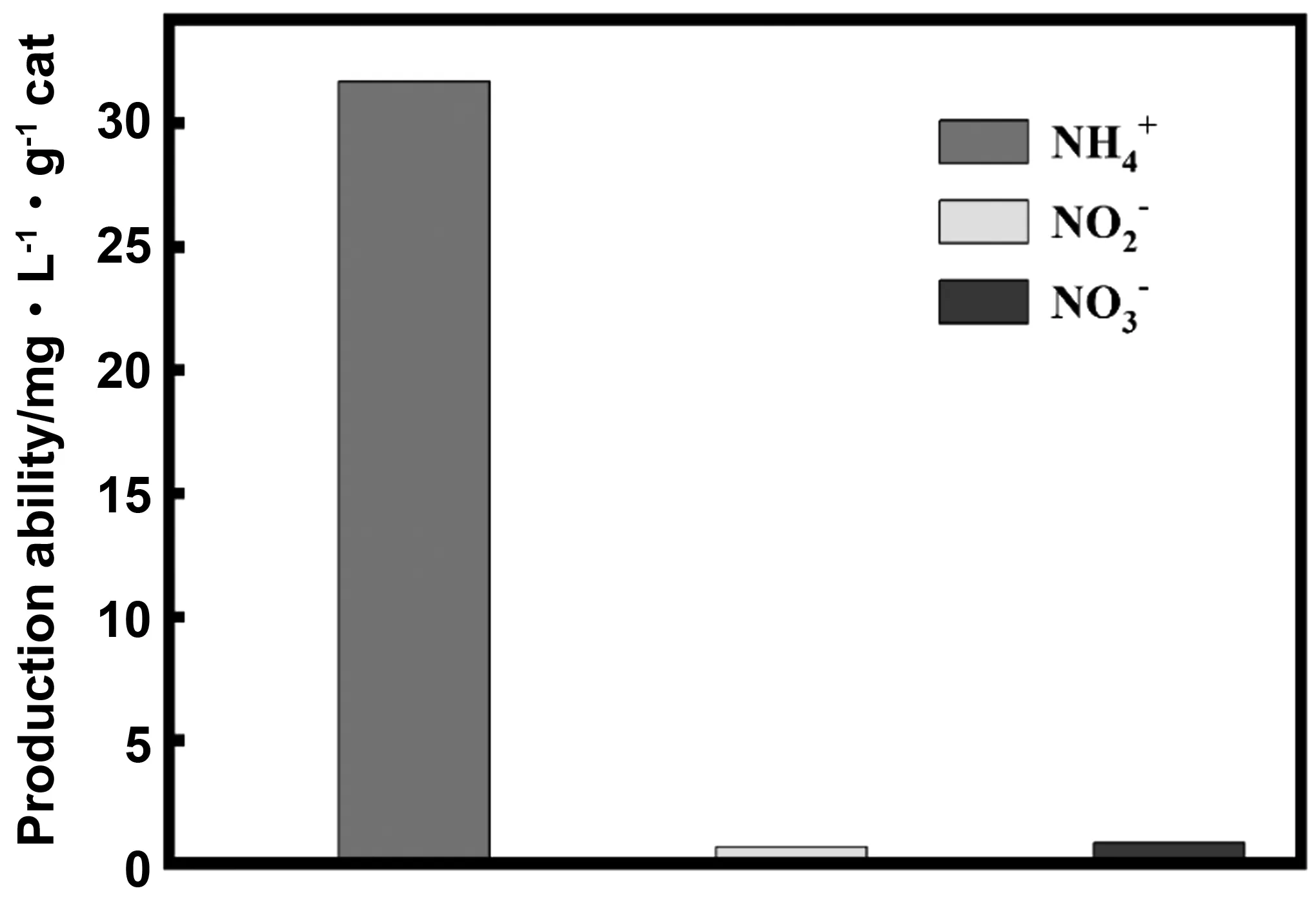
3.1 不同體系下光催化固氮的影響
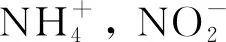
3.2 P摻雜量對光催化固氮影響
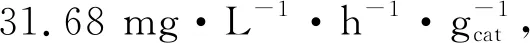

圖7 不同的光催化固氮體系
3.3 捕獲劑種類對光催化固氮效果的影響

圖和 的產量
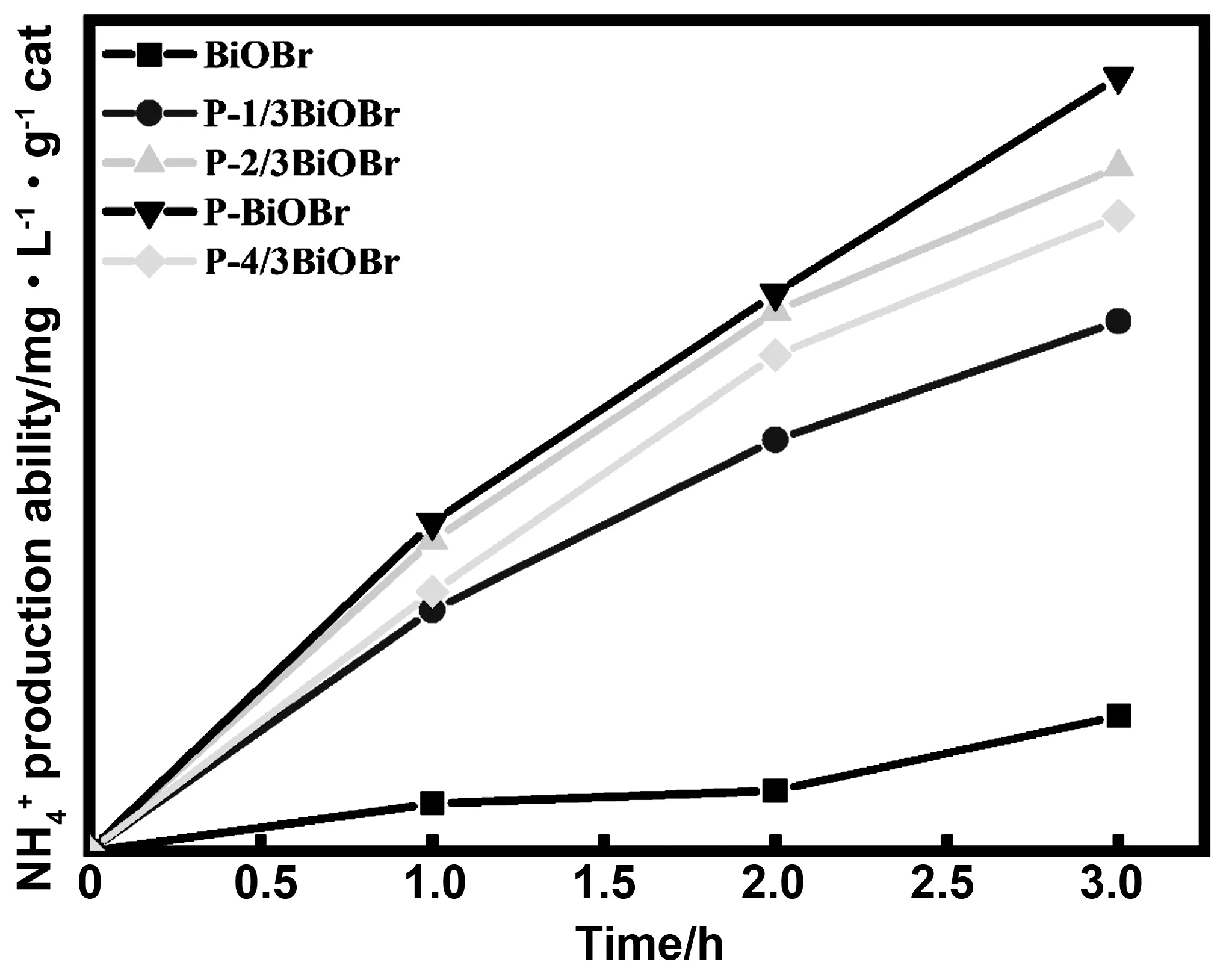
圖9 不同光催化劑的光催化固氮效果
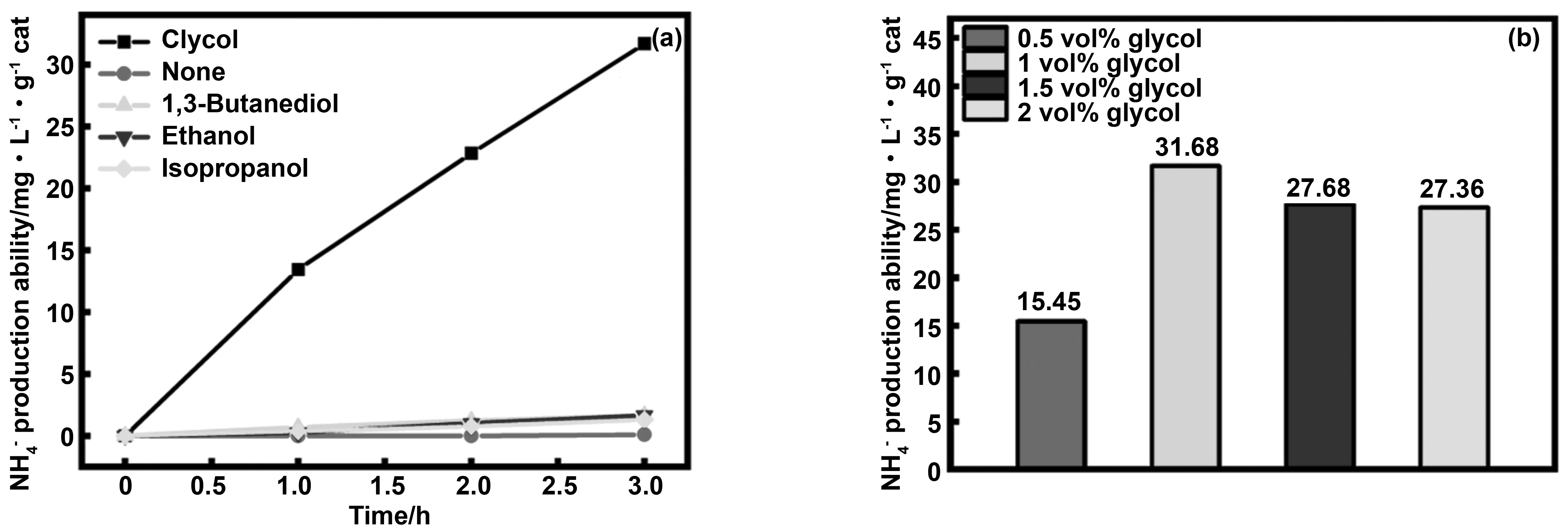
圖10 捕獲劑的種類和濃度對固氮效果的影響
3.4 光催化劑穩定性實驗
為了探究催化劑P-BiOBr的穩定性,將每一次固氮反應后的催化劑回收,再加入反應體系中繼續進行光催化固氮。如圖11所示,3次回收的固氮量與第一次固氮量沒有明顯的差別, P-BiOBr經過12 h的固氮反應仍然在繼續,這說明該催化劑有較好的穩定性和可循環性,具有很好的發展前景。
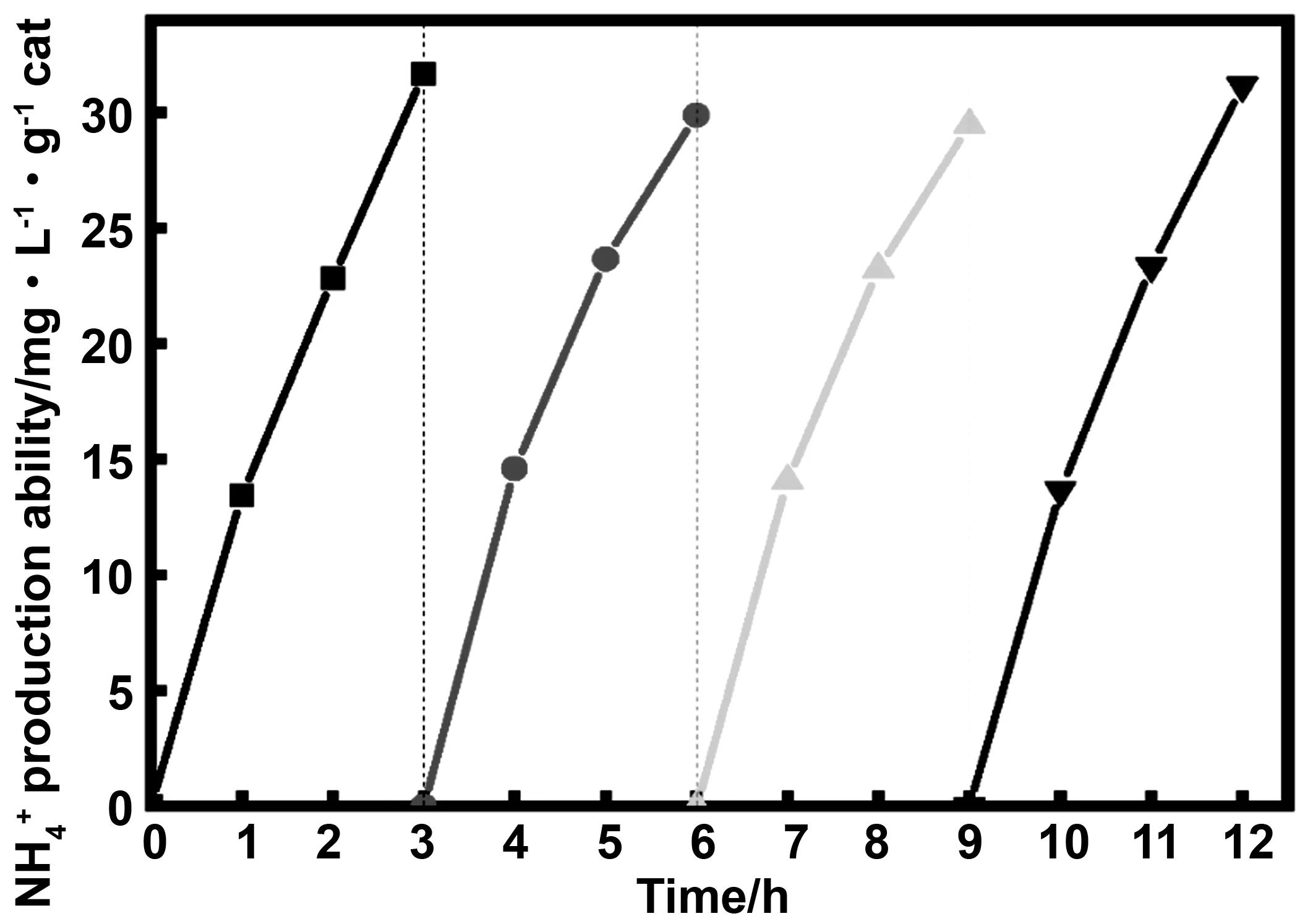
圖11 催化劑的循環實驗
4 結 論
(1)將次亞磷酸鈉作為P源,采用溶劑熱法,將P元素摻雜到BiOBr,發現不同摻雜量的催化劑的固氮效果均有提升,P元素摻雜可以提高固氮性能,其中以Bi,P摩爾比為15∶1的P-BiOBr固氮效果最佳,是BiOBr的4.7倍。
(2)P元素的摻雜使催化劑產生了雜質帶,提高BiOBr的表面電荷轉移效率,因此固氮效果有所提升。
(3)P-BiOBr增強了催化劑的瞬態光電流強度,并且P的摻雜可能產生微量的BiPO4,有助于增強載流子的光吸收和分離效率,進而提高了固氮效率。
(4)經過4次循環實驗,P-BiOBr的固氮效果沒有明顯改變,說明該材料穩定。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中老年保健(2021年12期)2021-11-30 02:58:01
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
攝影之友(影像視覺)(2019年2期)2019-03-05 08:27:14
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
中華詩詞(2018年11期)2018-03-26 06:41:34
Coco薇(2016年8期)2016-10-09 02:11:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
