Rosai-Dorfman病18例臨床病理特征
2022-12-21 02:49:58鄧雅婷王小偉彭宏凌
臨床與實驗病理學雜志 2022年11期
趙 艷,鄧雅婷,蔣 誼,王小偉,彭宏凌
Rosai-Dorfman病(Rosai-Dorfman disease, RDD)又稱竇組織細胞增生癥伴巨大淋巴結病,是一種可累及淋巴結和(或)結外部位、較少見的非朗格漢斯細胞組織細胞增生癥。該病由法國病理學家Destombes[1]于1965年首次描述,1969年Rosai和Dorfman[2]對患者臨床病理特點進行分析,將其命名為RDD。RDD主要發生于兒童和年輕人,通常表現為雙側、大而無痛的頸部淋巴結病,伴或不伴間歇性發熱、盜汗和體重減輕。據文獻報道,RDD結節外受累的發生率為43%,僅19%發生多系統受累[3]。目前,RDD首選手術治療,無法進行手術的患者可選用局部治療和系統治療。本文著重探討RDD的臨床病理學特征、診斷及鑒別診斷,以期提高臨床醫師對該疾病的認識水平。
1 材料與方法
1.1 材料收集2010年11月22日~2021年9月14日中南大學湘雅二醫院經病理確診的18例RDD,其中男性10例,女性8例,男女比為5 ∶4;年齡12~64歲,平均37.9歲,中位年齡42歲;病程10余天~1年余。10例患者以淋巴結腫大為初發癥狀,其中1例伴罕見的腮腺受累;3例患者自訴有外傷史;2例有結核病史;1例有黃疸病史;余2例未見特殊病史。臨床診斷曾考慮為RDD 3例,淋巴瘤3例,骨腫瘤2例,頸椎骨質破壞1例,梗阻性黃疸1例,腎腫瘤1例,結締組織病1例,反應性關節炎1例,抗合成酶抗體綜合征1例,結節病1例,其他診斷包括:成人Still綜合征、血管瘤、胸椎腫瘤各1例。
1.2 方法
1.2.1免疫組化 標本均經10%中性福爾馬林固定,常規脫水,石蠟包埋,4 μm厚連續切片,采用HE、免疫組化EnVision兩步法染色。一抗S-100、CD68、vimentin、CD1a、Langerin、Ki-67等及二抗,均購自北京中杉金橋公司。結果判斷采用半定量分析:陽性細胞數<10%為陰性(-),10%~30%為弱陽性(+),>30%~75%為中度陽性(),>75%為強陽性()。
1.2.2原位雜交 EBER原位雜交檢測EBER-1探針試劑盒,購自北京中杉金橋公司,具體操作步驟嚴格按試劑盒說明書進行;以細胞核內出現藍紫色顆粒為陽性信號。判斷標準:陽性細胞數<5%為陰性(-),5%~25%為弱陽性(+),>25%~50%為中度陽性(),>50%為強陽性()。
2 結果
2.1 臨床特征本組RDD患者9例為單發(50%),9例為多發(50%)。18例患者中5例因淋巴結腫大就診,6例因腫塊形成(包括面部、下肢、肋骨、肱骨、脊椎、肩部、胸背部)就診,2例因發熱、皮疹伴關節痛就診,1例因頸椎病理性骨折就診,1例因黃疸就診,1例因無痛肉眼血尿就診,1例因腮腺腫塊就診,1例因頭痛就診。發病部位:10例位于淋巴結,6例位于骨關節,3例位于皮膚,2例位于脊椎,2例位于臟器(肝與腎),2例位于軟組織(肩部/胸背部),2例位于眼,1例位于中樞神經系統,1例位于腮腺。18例患者臨床表現多樣:發熱6例、咳嗽咳痰3例、瘙癢7例、淋巴結腫大10例、皮膚病變4例、頭痛1例、關節痛6例、眼睛紅腫2例等。18例患者從發病至確診的平均時間約為3.4個月。RDD初診實驗室相關指標:乳酸脫氫酶指標異常占比60%(6/10);貧血為50%(9/18);血小板數異常為44.4%(8/18);D-Dmier和白蛋白量指標異常均為33.3%(6/18);外周血白細胞和中性粒細胞指標異常均為27.8%(5/18)。
2.2 影像學特征18例患者中有8例完善PET-CT檢查,10例行頸部、胸腹部及骨骼CT檢查,6例行MRI檢查。PET-CT檢查:患者全身多處廣泛增大的淋巴結,并伴糖代謝不同程度增高。5例出現脾大,伴糖代謝增高;6例全身骨骼糖代謝彌漫性增高,提示骨髓增生活躍。1例累及肋骨,導致骨質破壞,PET-CT受累部位最大SUV為7.8;提示淋巴結和骨組織是RDD患者最常見的受累部位。CT、MRI等檢查均發現多部位疑似淋巴結腫大或軟組織增生影。
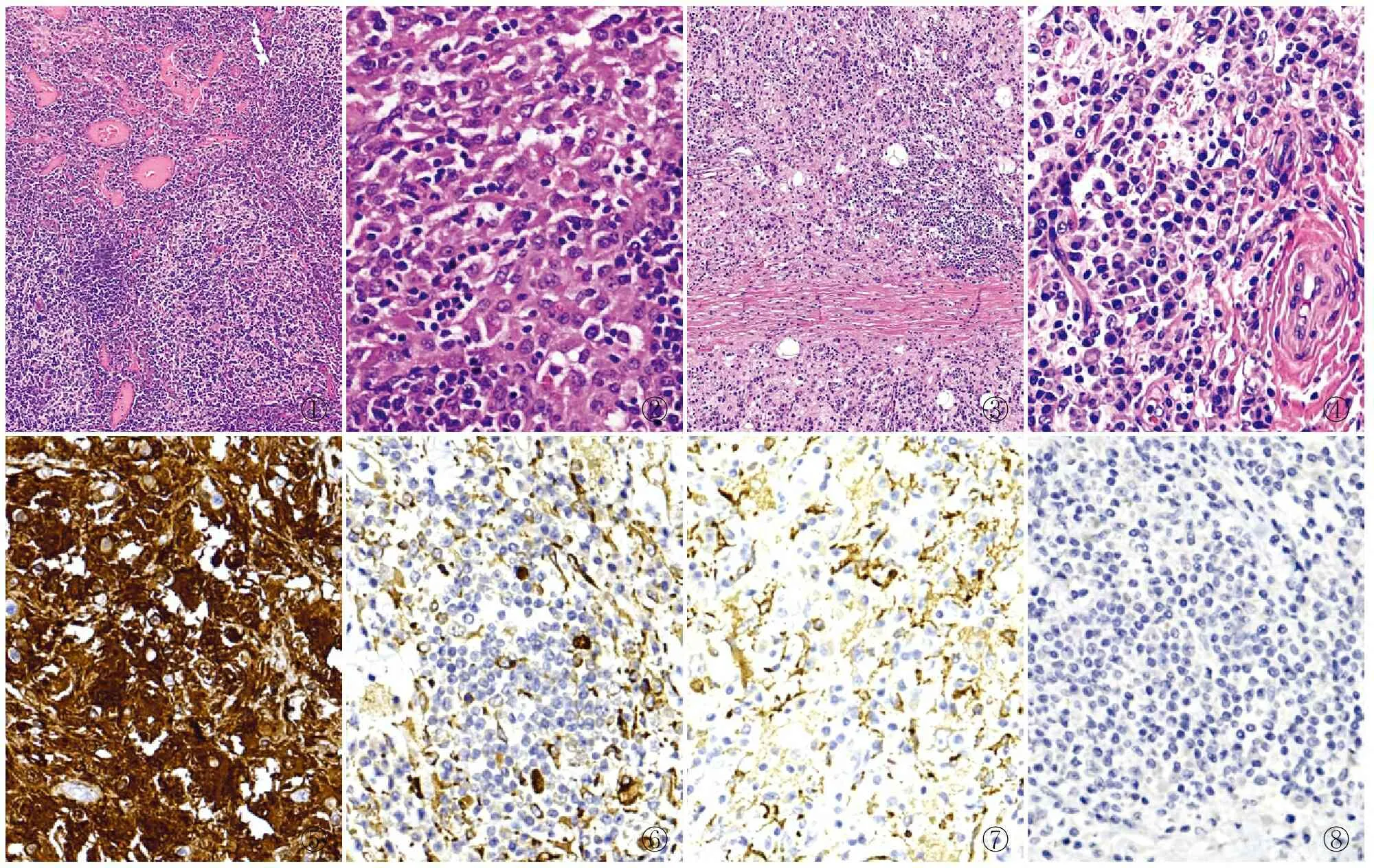
2.3 病理特征眼觀:病變部位邊界欠清,皮膚型表現為暗紅色腫塊。腫物切開均為實性、切面灰白色,質地較硬。最大徑0.2~7.5 cm,部分有包膜。鏡檢:低倍鏡下病變區域呈“明暗相間”不規則分布;高倍鏡下明區可見組織細胞增生,呈結節狀分布,細胞呈卵圓形或多邊形,胞質豐富,淡染或嗜伊紅色,細胞有一定的異型性,核仁明顯,見個別雙核細胞;部分組織細胞呈大小不一的空泡狀。18例患者胞質內可見被吞噬的淋巴細胞及漿細胞,其中3例還可見吞噬紅細胞,偶可見中性粒細胞。暗區可見大量淋巴細胞和漿細胞聚集;炎性背景均見大量間質纖維化及膠原化。淋巴結型RDD:鏡下見多數淋巴結結構基本存在,部分組織細胞胞質內有淋巴濾泡,淋巴竇明顯擴張,竇內組織細胞增生,部分組織細胞質內有淋巴細胞吞噬現象(即“伸入運動”)(圖1、2);皮膚型RDD:真皮層及皮下脂肪組織等受累,鏡下見病變組織內大量淋巴細胞、漿細胞及組織細胞浸潤,淋巴濾泡形成,可見“伸入運動”(圖3、4)。
2.4 免疫表型及原位雜交18例RDD組織中S-100(圖5)、CD68(圖6)、CD163(圖7)、vimentin、CD21均陽性,CD1a(圖8)、Langerin、CK、CD10、CD15、CD30、HMB-45均陰性。2例CD34血管陽性;5例CD45陽性。漿細胞中CD38、CD138、MUM1均陽性。淋巴細胞中CD3、CD20均陽性。T細胞中CD4、CD8、CD45均陽性。B細胞中CD20陽性。3例IgG4陽性,其中1例IgG陰性,1例評估IgG4/IgG陽性比>40%,故診斷傾向伴IgG4相關性疾病的皮膚型RDD。15例RDD行Ki-67增殖指數2%~80%,中位數為20%(<40%),余3例未檢測;其中1例患者Ki-67增殖指數80%,為淋巴瘤伴RDD。本組8例患者EBER原位雜交檢測陰性,9例未檢測,1例伴T細胞非霍奇金淋巴瘤患者EBV陽性。5例行Kappa、Lambda染色均為雙陽性,考慮為單克隆可能性大。
2.5 治療及預后本組18例患者中9例行手術切除,6例行激素(如潑尼松)治療,同時輔以免疫調節、抗感染等治療,無癥狀者給予一般治療。本組9例手術治療患者中有8例預后良好,未復發,僅1例病情嚴重,需經多次手術治療。余通過激素治療等部分患者淋巴結大小和癥狀得到改善。
3 討論
RDD屬于少見的良性組織細胞增生性疾病[4],在富含淋巴細胞、漿細胞和中性粒細胞的多形性炎癥細胞背景中具有巨噬細胞增殖特性。RDD被認為是病因不明的自限性疾病,可能與免疫功能缺陷、細胞因子調節異常(IgG4)或病毒感染有關[5]。其典型的病理特征為:S-100與CD68陽性,CD1a陰性。
3.1 臨床特點RDD臨床表現為雙側無痛性頸部淋巴結腫大,可累及縱隔、腋窩和腹股溝淋巴結。實驗室可見紅細胞沉降率升高,白細胞增多,高球蛋白血癥和自身免疫性溶血性貧血。此外,RDD最常見的結外受累部位是皮膚,約占RDD患者總數的10%。馮肖等[6]報道,結外主要發生于頭頸部。其他部位如中樞神經系統、骨、眼眶和眼瞼也有受累現象。RDD好發于兒童和年輕人,平均發病年齡為20.6歲,男性多見;皮膚型患者發病年齡較大,以女性為主[7]。本組患者臨床表現與文獻相符,但發病年齡較大,中位年齡42歲,最大者發病年齡達64歲。
3.2 病理特征淋巴結型RDD:(1)早期鏡下見多數淋巴結結構基本存在,內有淋巴濾泡,擴張的淋巴竇內組織細胞增生。部分組織細胞胞質內有數量不一、形態完整的淋巴細胞、漿細胞及中性粒細胞,稱為“伸入運動”,是具診斷性的病理表現。(2)晚期患者淋巴結結構消失,組織細胞彌漫性浸潤。皮質中存在大量活化的B細胞和成熟漿細胞,其與蒼白的組織細胞呈明暗交替的外觀。與淋巴結型RDD不同的是,皮膚型RDD表現為更多的淋巴濾泡,伴生發中心、纖維化、硬化、更少的組織細胞和更細微的結締組織增生。本組18例RDD多為多發型,均有“伸入運動”并伴不同程度的纖維化。
①②③④⑤⑥⑦⑧
3.3 免疫表型多數RDD組織細胞中S-100、CD68、CD163和CD14均陽性,且CD1a和Langerin陰性有助于明確診斷。S-100染色通常會突顯出“伸入運動”的特點,與本實驗相符。
3.4 鑒別診斷RDD診斷依據臨床特點及形態學改變,免疫組化染色可進行排除性診斷。(1)朗格漢斯細胞組織細胞增生癥:是一種原因未明的組織細胞異常增殖伴炎癥浸潤引起的惡性腫瘤[8],朗格漢斯細胞組織細胞增生癥Langerin多異常陽性,且無“伸入運動”。本組S-100、CD68和CD163均陽性,與朗格漢斯細胞組織細胞增生癥相比,RDD細胞中CD1a/Langerin陰性。(2)Erdheim-Chester病:屬于罕見的非朗格漢斯細胞組織細胞增生癥,亦稱為黃色肉芽腫病,好發于中老年人,50%~100%的患者攜帶BRAF V600E突變[9]。有研究報道,Erdheim-Chester病中S-100(20%~30%)可陽性,臨床最常累及骨骼系統(84.6%),其次是中樞神經系統(46.2%)和眼部(30.8%)。當中樞性尿崩癥伴發熱、骨痛或多器官系統受累時,需考慮Erdheim-Chester病的可能。(3)T細胞非霍奇金淋巴瘤:主要起源于淋巴結或結外淋巴組織,為T淋巴細胞來源的惡性增殖性腫瘤,主要表現為無痛性淺表淋巴結腫大,呈進行性發展,以頸部最為常見。患者常伴腹部包塊、上腹痛、消化道出血等表現,近些年其發生率明顯升高[10]。多因感染人T細胞白血病病毒-1(human T-cell leukemia virus, HTLV-1)和EBV引起。臨床曾有1例誤診,進一步行T淋巴細胞基因重排檢測陽性可確診。(4)IgG4相關性疾病:其特點為類腫瘤樣病變、血清IgG4水平升高、IgG4陽性漿細胞浸潤及組織纖維化[11-12]。與RDD組織學特征較相似,且RDD可伴隨IgG4和IgG陽性的漿細胞存在。本組有1例伴IgG4相關性疾病的皮膚型RDD,診斷標準為IgG4/IgG陽性比值>40%。
3.5 影像學檢查RDD根據累及器官及部位的不同,影像學特征有所差異。具體表現與多種腫瘤和炎性疾病有重疊[13]。本組CT顯示RDD為孤立或播散性淋巴結病,單發或多發,增強掃描可呈均勻強化。全身MRI可代替CT,以避免兒童和年輕患者輻射,通常表現為淋巴結腫大或軟組織增生。PET/CT可提示全身多處廣泛增大的淋巴結,可用于疾病的初步評估[14]。在受累部位可見大量示蹤劑攝取,隨著治療的成功,示蹤劑的攝入減少[15]。
3.6 治療及預后RDD臨床過程及結局通常是良性且自限,目前尚無標準的治療方案。RDD的治療包括手術、放、化療、類固醇、免疫抑制劑沙利度胺或干擾素α、利妥昔單抗和靶向治療。無癥狀者可給予一般治療;在活檢、皮膚RDD、單個病灶疾病、壓迫或阻塞的情況下可能需要行手術治療[16];為改善淋巴結腫大癥狀時可以選擇激素療法;在難治性或復發性病例中,化療可以緩解患者病情。當RDD患者出現KRAS、MAP2K1等激酶突變或BRAF V600E等基因突變時,可行cobimetinib[17]、BRAF抑制劑dabrafenib[18]等靶向治療。累及多系統的臨床表現、慢性病程和潛在的免疫紊亂常出現于年齡較大的老年患者,其預后較差。同樣,結外受累時預后較差,可能持續存在。少數病例病程持久,或因多器官侵犯而死亡。本組中9例患者行手術治療,8例預后較好,未見復發;1例患者癥狀較重,多次手術后病情好轉。
綜上所述,RDD是臨床少見的非朗格漢斯細胞組織細胞增多癥,易誤、漏診;主要臨床表現為雙側、大而無痛的頸部淋巴結腫大,最常見的部位是淋巴結,也有結外受累現象。S-100、CD68、CD1a、Langerin是RDD重要的病理診斷依據,需與其他疾病進行鑒別;其根據病變累及器官組織部位的不同影像學表現存在差異。RDD診斷應包括仔細分析病理學特征、臨床表現和支持性影像學表現。RDD臨床過程通常具有自限性,預后較好。因此,可根據患者病情進行個性化治療。
