MDM4 rs4245739基因多態性與乳腺癌易感性的meta分析
2023-01-05 03:19:08李佳陽伍紅瑜陳保林程曉明呂俊遠
臨床薈萃 2022年11期
姜 焱,李佳陽,伍紅瑜,陳保林,程曉明,呂俊遠
(1. 遵義醫科大學附屬醫院 a. 普通外科; b. 乳腺甲狀腺外科; c. 藥物臨床試驗機構, 貴州 遵義 563099; 2. 遵義醫科大學 基礎藥理學教育部重點實驗室暨民族藥教育部國際聯合研究實驗室,貴州 遵義563000)
乳腺癌是女性最常見的惡性腫瘤之一,約占女性惡性腫瘤的23%,也是導致女性癌癥相關性死亡的主要原因,全球每年有超過60萬人死于乳腺癌[1]。乳腺癌的發生發展通常是由遺傳易感性和環境暴露之間復雜的相互作用而引起的[2-3]。越來越多的研究表明,遺傳因素在癌癥的發病機制中發揮重要作用,且目前有許多基因已被鑒定為癌癥易感基因[4]。
鼠雙微體4(mouse double minute 4,MDM4)基因位于染色體1q32,該基因編碼一種核蛋白,在N端含有腫瘤抑制基因p53的結合域,其可通過結合轉錄激活域抑制p53,進而參與乳腺癌的發生發展[5]。單核苷酸多態性(single nucleotide polymorphism, SNP)可影響基因和蛋白水平的穩定性[6],rs424539是MDM4基因目前研究較多的SNP位點,許多研究已表明MDM4rs4245739的基因多態性可影響MDM4的表達進而影響乳腺癌發生發展進程。Liu等[7]的研究發現MDM4rs4245739的基因多態性可降低乳腺癌發生的易感性;而相反地,Hashemi等[8]的研究表明乳腺癌易感性與MDM4rs4245739的基因多態性無關;而Wang等[9]通過meta分析發現,MDM4rs4245739的基因多態性與亞洲人乳腺癌易感性呈負相關,但該meta分析納入研究不全,未將Montserrat 等[10]的研究納入。因此,本研究通過meta分析現有研究,旨在探討MDM4rs4245739的基因多態性與乳腺癌易感性的相關性。
1 資料與方法
1.1檢索策略 我們在PubMed、EMBASE、Cochrane Library、中國知識基礎設施數據庫和萬方電子數據庫中檢索相關文獻。以下所有的關鍵詞均在各個數據庫中檢索:“乳腺癌”、“乳腺腫瘤”、“breast cancer”、“breast tumor”、“breast malignance”、“鼠雙微體4”、“MDM4”、“HDMX”、“MDMX”、“MRP1”、“單核苷酸多態性”、“基因多態性”、“基因亞型”、“基因突變”、“基因變異”、“single nucleotide polymorphism”、“SNP”、“gene polymorphism”、“gene subtype”、“gene mutation”、“gene variation”。此外,還對合格文獻的參考文獻和相似文獻列表進行了審查,以確保檢索到所有可能合格的文獻。
1.2納入標準 (1)病例-對照研究; (2)關于MDM4基因多態性與乳腺癌易感性的相關性研究;(3)可獲取全文; (4)有足夠的基因型分布數據,可用于計算OR和95%CI;(5)健康對照者人群的基因型分布必須符合Hardy-Weinberg平衡(Hardy-Weinberg equilibrium,HWE) 。
1.3排除標準 (1)無健康對照者的研究;(2)無詳細乳腺癌患者數、健康對照者數和一些其他重要信息;(3)排除綜述和數據重復文獻。
1.4數據提取和質量評估 對所有符合條件的納入文獻,提取了以下數據:作者、發表年份、地區、種族、樣本量以及病例和健康對照者的基因型頻率。兩位作者獨立提取數據,并基于修正的紐卡斯爾-渥太華量表(Newcastle-Ottawa Scale,NOS)評估納入文獻的質量[11]。NOS是一種半定量的評分表,總分從0分(最低質量)到9分(最高質量),評分為5~9分表示高質量。兩名獨立的作者對這些研究進行了獨立的評分,如果對同一篇文章的評分結果不同,則通過討論解決評分的分歧。
1.5統計學方法 使用Stata 15.1軟件(Stata Corporation, University Station, Texas, USA)對所有相關數據進行統計分析。利用卡方檢驗,計算每個病例和健康對照者的等位基因和基因型頻率,以評估HWE值。通過OR和95% CI估計在等位基因模型(C vs A)、顯性基因模型(AC+CC vs AA)、隱性基因模型(AC+AA vs CC)、雜合子基因模型(AC vs AA)、純合基因模型(CC vs AA)下的MDM4rs4245739基因多態性與乳腺癌易感性之間的關聯強度。異質性檢驗采用Q檢驗,用I2進行量化,當P>0.05或I2<50%時表示異質性較小,則采用固定效應模型。相反,則選擇隨機效應模型[12]。當發現有統計學上的異質性時,進行亞組分析和meta回歸分析,探討研究中可能的異質性來源。我們還通過逐一排除法進行了敏感性分析,以確定每個研究對總體異質性的影響,以評估總體結果的穩定性。采用Begg's漏斗圖和Egger's線性回歸來評估可能的發表偏倚[13]。P<0.05表示差異有統計學意義。
2 結 果
2.1納入研究的特征 經過初步檢索,總共確定了289篇文獻。通過剔除重復、篩選標題和摘要后排除272篇文獻。其中保留了17項研究進行全文閱讀和審查,另有12篇研究因下列原因被排除:系統性綜述文章4篇[9, 14-16];關于非rs4245739位點基因多態性的研究2篇[17-18];無健康對照者6篇[19-24]。最后,有5篇文獻符合納入標準被納入本meta分析,涉及9 814名乳腺癌患者和45 202名健康對照者[7, 8, 10, 25-26],見圖1。在健康對照者的基因型分布中,所有研究都與HWE一致。所有納入研究的特征見表1。
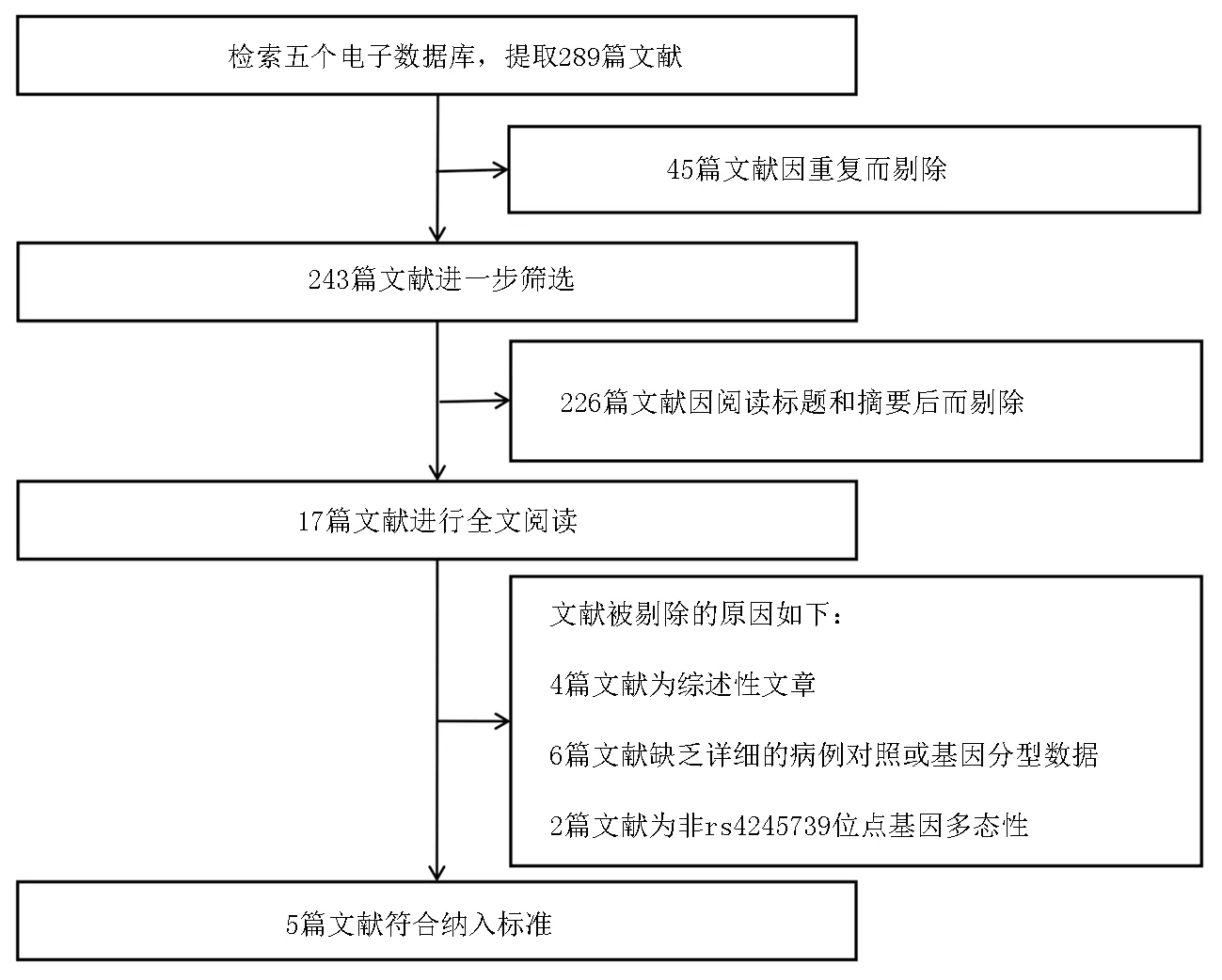
圖1 文獻篩選流程圖
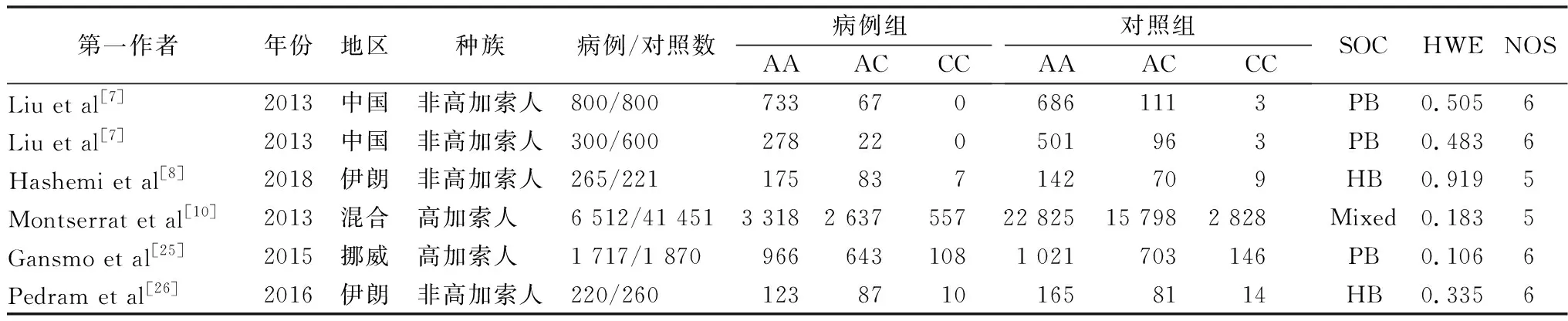
表1 納入文獻的基本特征
2.2Meta分析結果 本研究共納入5篇病例-對照文獻(含6項研究)來匯總數據分析MDM4rs4245739多態性與乳腺癌風險相關性。結果顯示,MDM4rs4245739基因多態性與乳腺癌易感性在任何遺傳模型中都沒有發現顯著的相關性:等位基因模型 (C vs A:OR=0.84, 95%CI: 0.67-1.05,P=0.118),顯性基因模型 (AC+CC vs AA:OR=0.86, 95%CI: 0.67-1.11,P=0.245),隱性基因模型(AC+AA vs CC:OR=0.90, 95%CI: 0.61-1.32,P=0.585),雜合子基因模型 (AC vs AA:OR=0.88, 95%CI: 0.69-1.12,P=0.305),純合子基因模型(CC vs AA:OR=0.90, 95%CI: 0.59-1.39,P=0.649),見圖2~6。
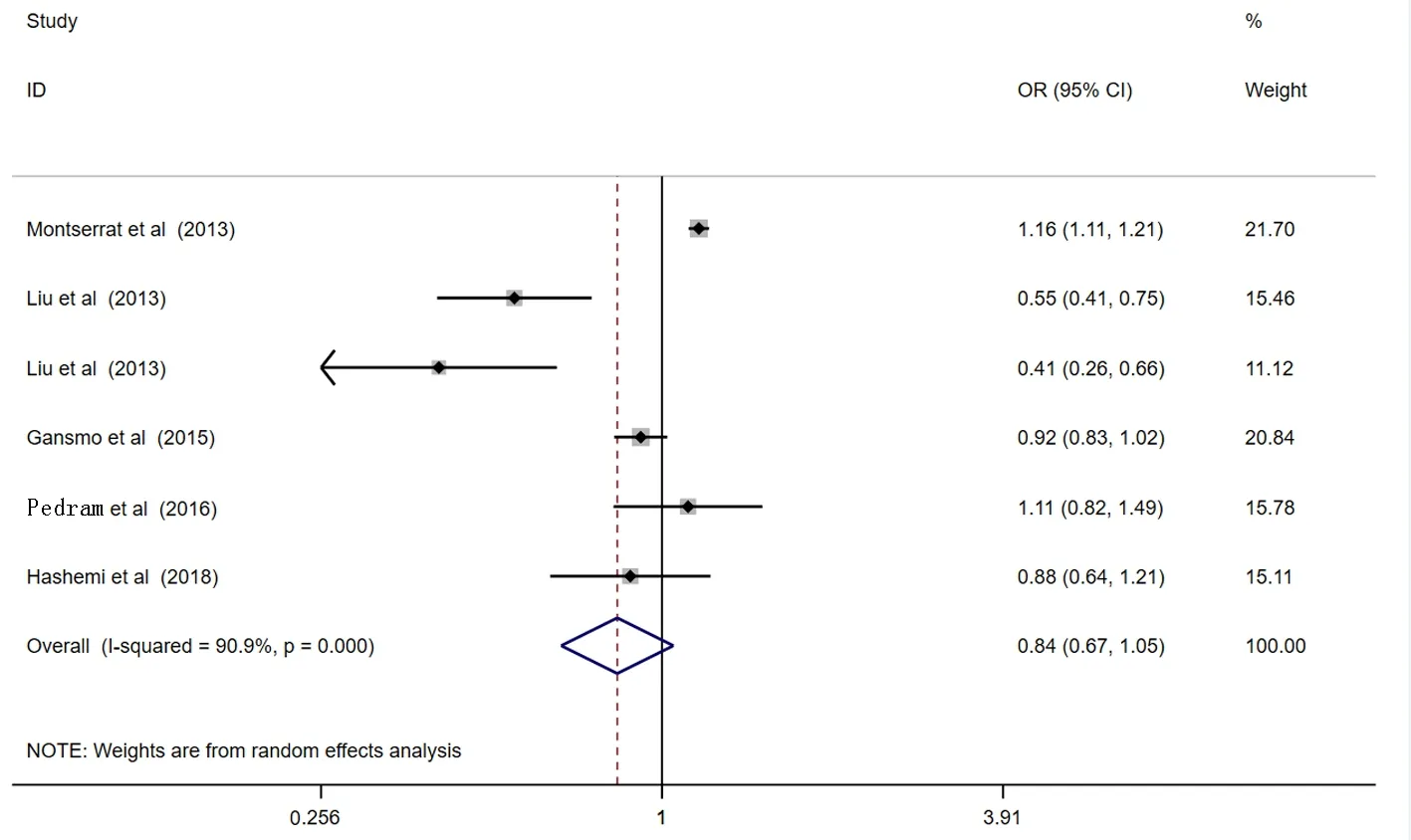
圖2 等位基因模型(C vs A)的森林圖

圖3 顯性基因模型(AC+CC vs AA)的森林圖

圖4 隱性基因模型(AC+AA vs CC)的森林圖
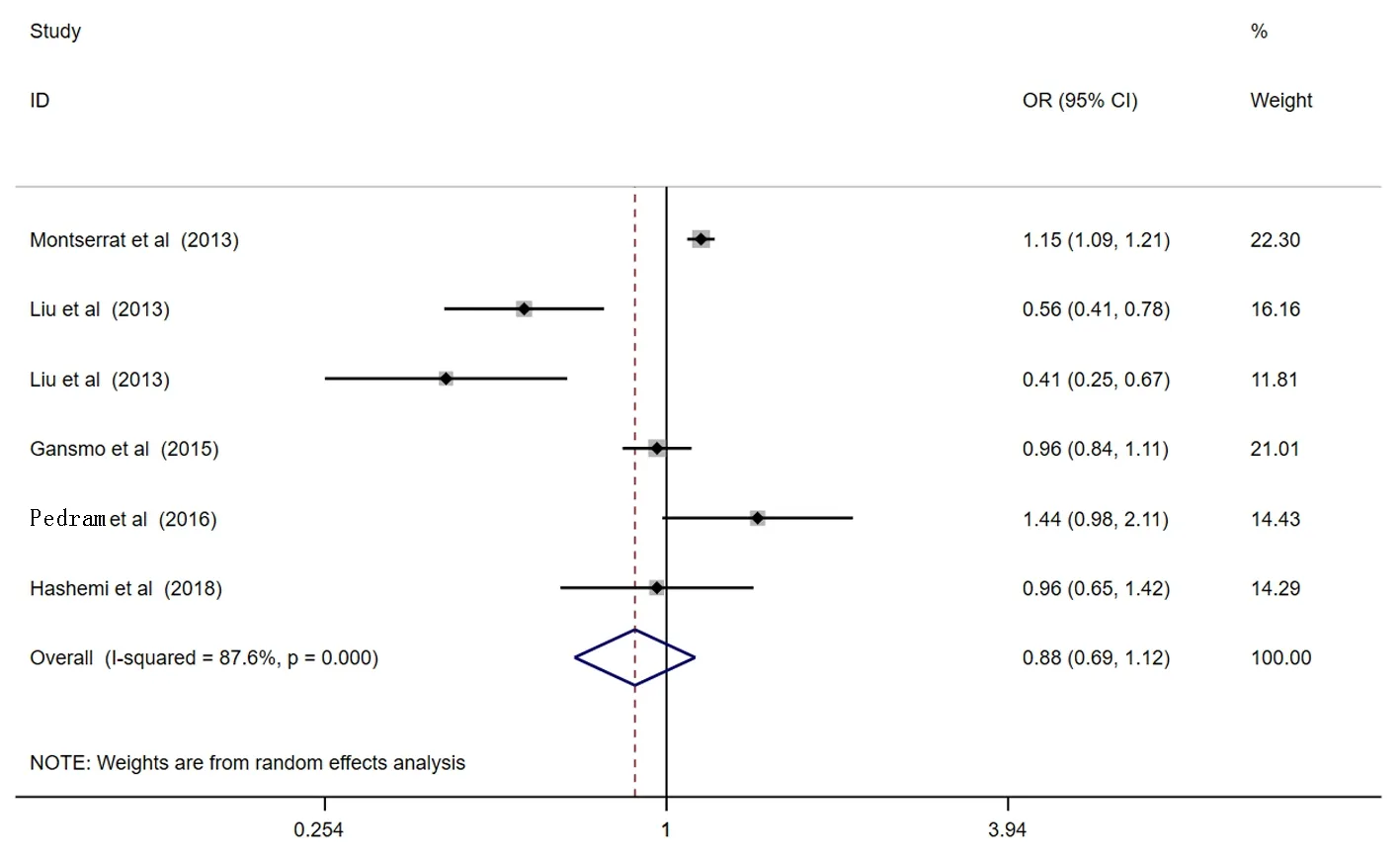
圖5 雜合子基因模型(AC vs AA)的森林圖
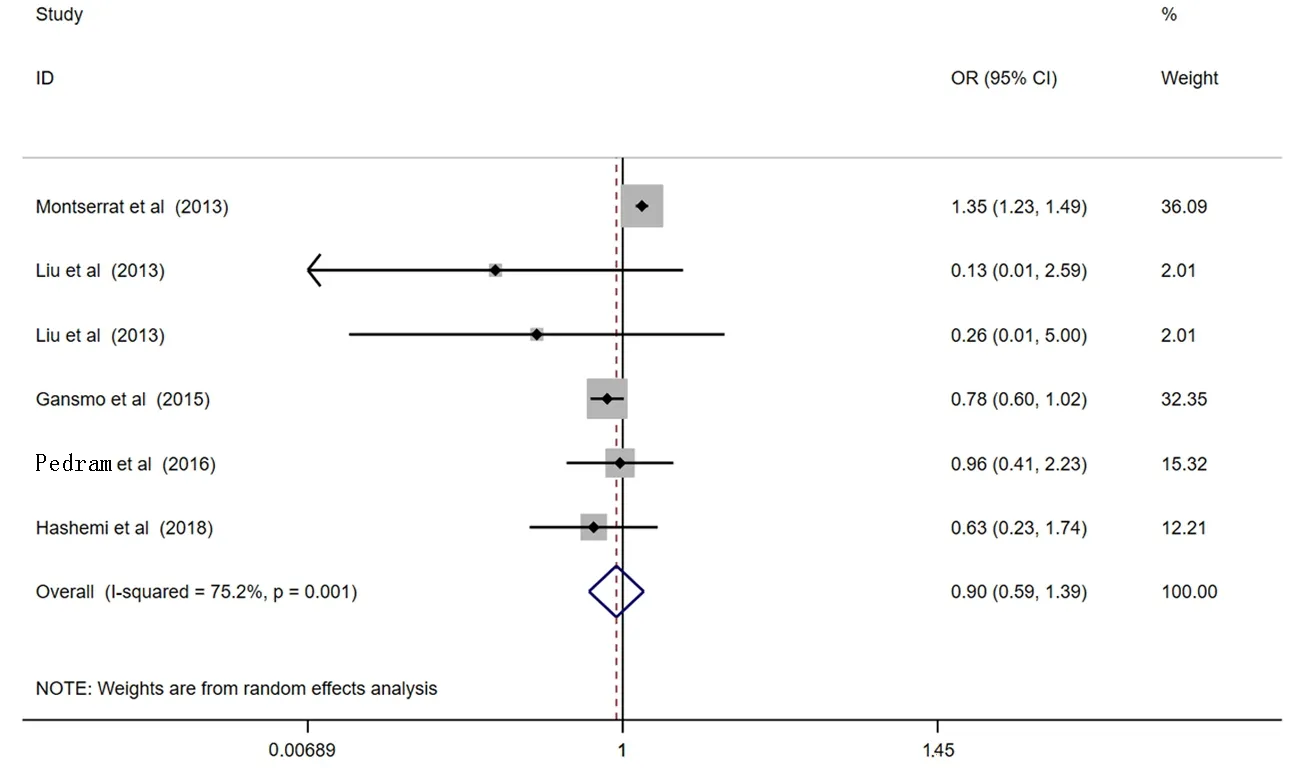
圖6 純合子基因模型(CC vs AA)的森林圖
2.3異質性檢驗 異質性檢驗結果顯示MDM4rs4245739 的5種基因模型間均存在顯著差異性:等位基因模型 (C vs A:I2=90.9%,P=0.000),顯性基因模型 (AC+CC vs AA:I2=90.0%,P=0.000),隱性基因模型 (AC+AA vs CC:I2=70.0%,P=0.005),雜合子基因模型 (AC vs AA:I2=87.6%,P=0.000),純合子基因模型 (CC vs AA:I2=75.2%,P=0.001)。因此,采用隨機效應模型來分析數據。為了闡明異質性的來源,我們進行了亞組和meta回歸分析。亞組分析和meta回歸結果顯示,健康對照者的地區分布和來源可能是導致異質性的主要來源,見圖2~6,表2~3。
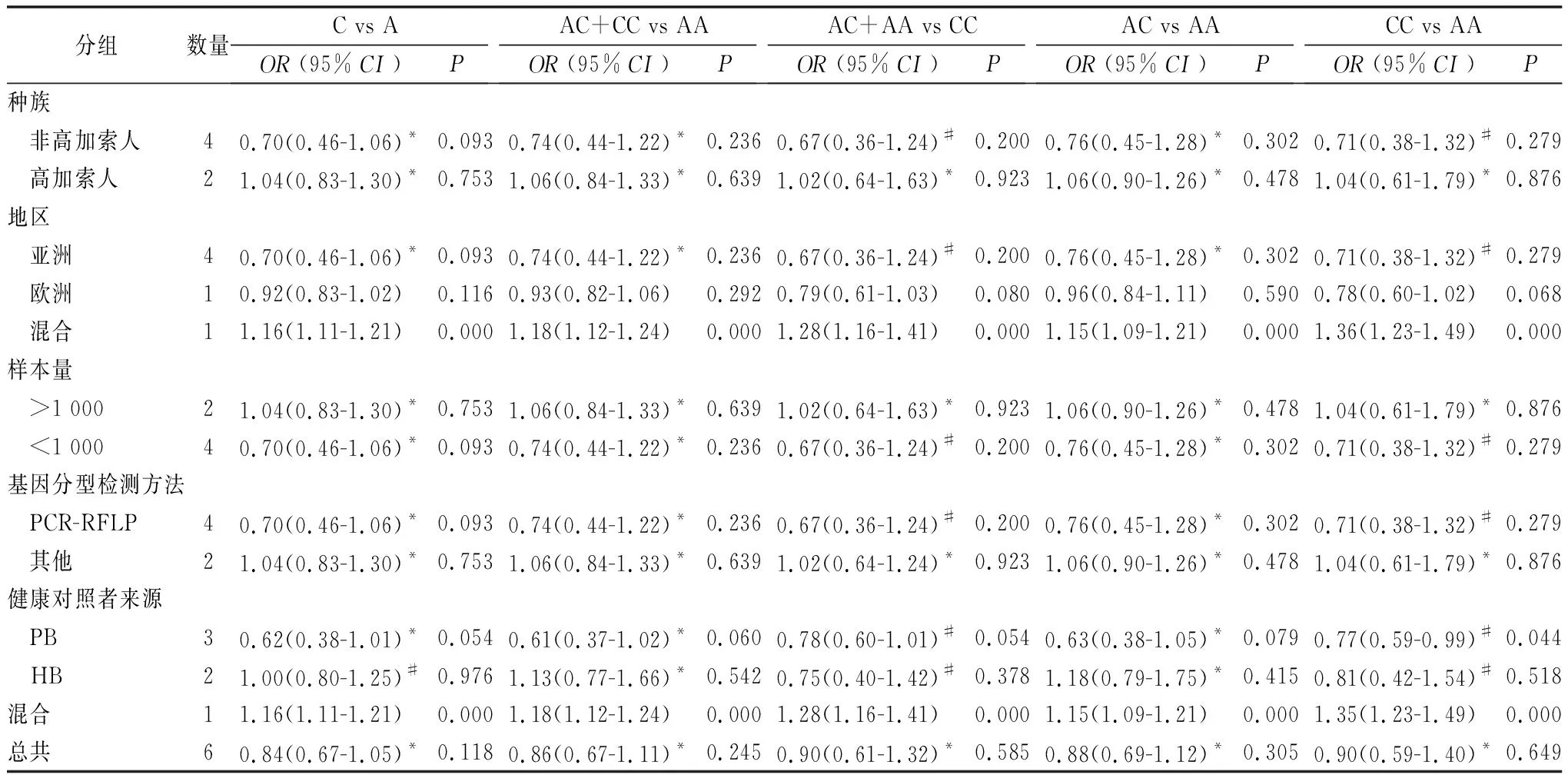
表2 亞組分析結果
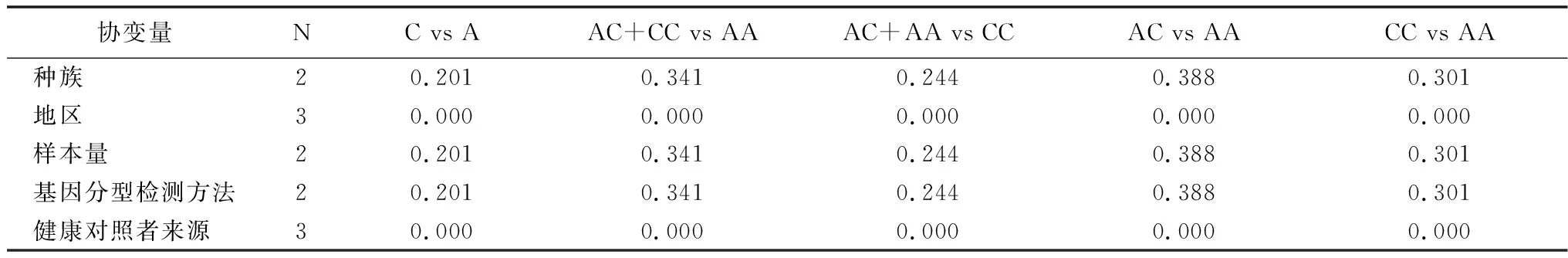
表3 Meta回歸結果
2.4敏感性分析 敏感性分析采用逐一排除法來評估每篇文獻對meta分析總體結果的影響。結果表明,在排除Montserrat等[10]研究后結果發生了改變,提示研究結果會受到該研究的影響,見圖7。
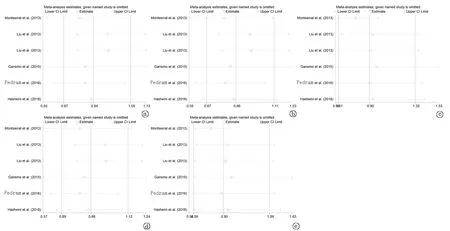
圖7 5種基因模型的敏感性分析 a. C vs A; b. AC+CC vs AA; c. AC+AA vs CC; d. AC vs AA; e. CC vs AA
2.5發表偏倚 采用Begg’s漏斗圖和Egger’s線性回歸檢驗來評估發表偏倚。漏斗圖存在不對稱性,提示發表偏倚可能。為此我們進一步使用Egger’s線性回歸檢驗評估發表偏倚風險,結果提示:等位基因模型(C vs A: PBegg’s=0.260, PEgger’s=0.044),顯性基因模型(AC+CC vs AA: PBegg’s=0.452, PEgger’s=0.100),隱性基因模型(AC+AA vs CC: PBegg’s=0.707, PEgger’s=0. 069),雜合子基因模型(AC vs AA: PBegg’s=0.452, PEgger’s=0.143) 和純合子基因模型(CC vs AA: PBegg’s=0.707, PEgger’s=0.090),見圖8。
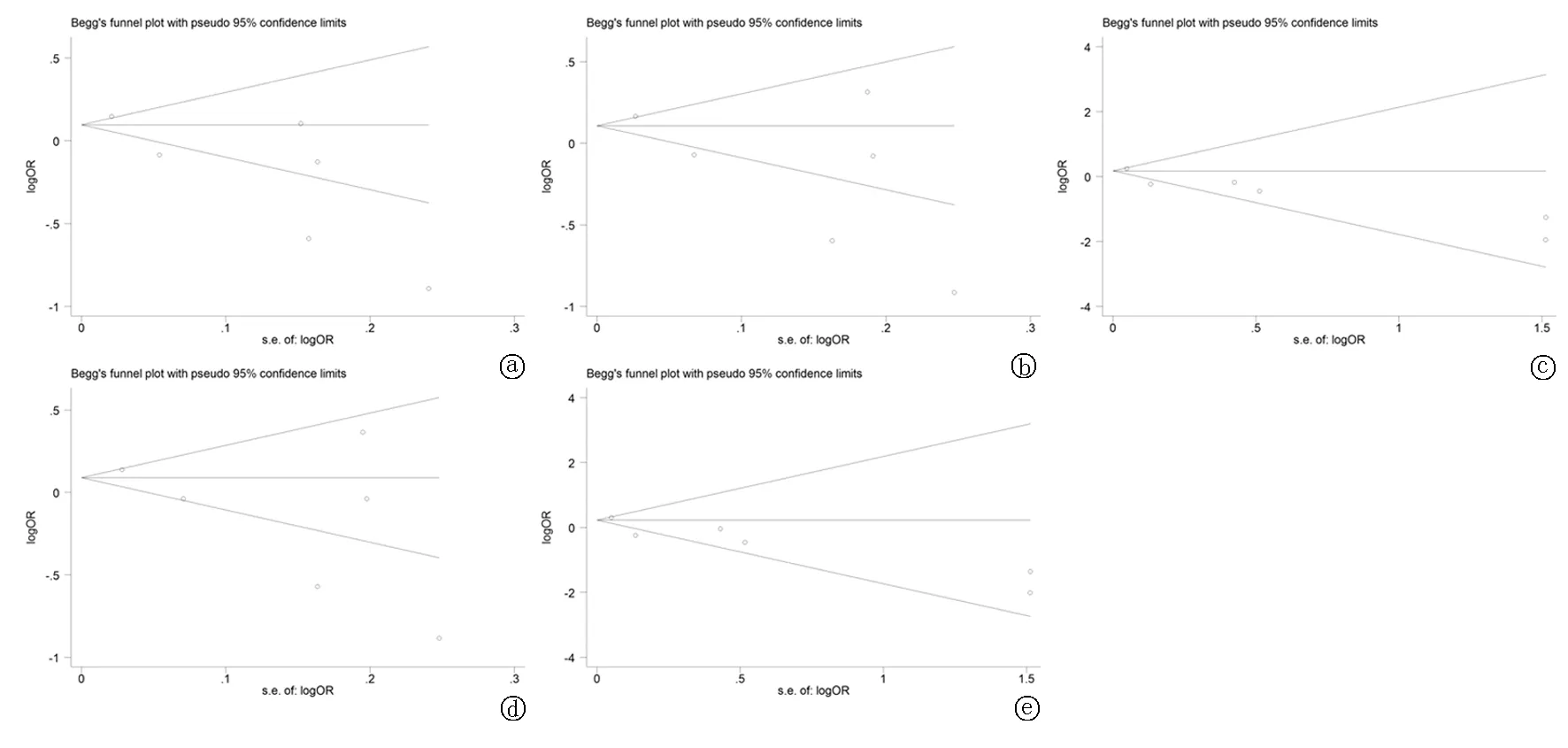
圖8 5種基因模型發表偏倚檢驗的Begg's漏斗圖 a. C vs A; b. AC+CC vs AA; c. AC+AA vs CC; d. AC vs AA; e CC vs AA
2.6GEPIA數據庫分析結果 本數據庫包含1 085例乳腺癌組織和291例正常乳腺組織樣本,分析結果顯示MDM4的表達水平與正常對照者差異無統計學意義,與乳腺癌的腫瘤分期和預后無關,見圖9。
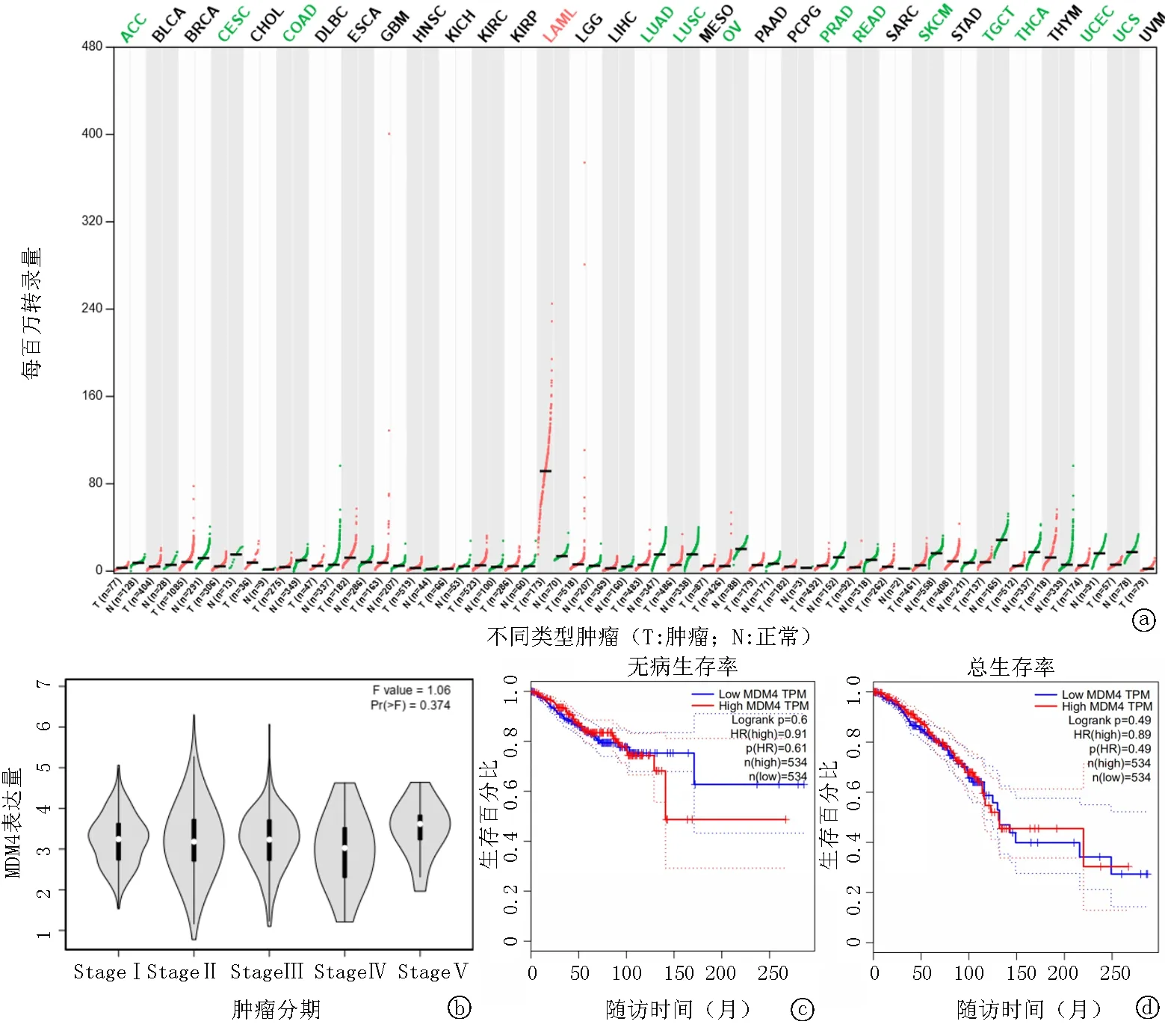
圖9 GEPIA數據庫分析圖 a. MDM4在不同類型腫瘤中的表達情況; b. MDM4在乳腺癌不同臨床分期中的表達情況; c. 乳腺癌患者MDM4表達與無病生存率的關系; d 乳腺癌患者MDM4表達與總生存率的關系
3 討 論
盡管分子生物研究提示MDM4可以通過調控p53參與腫瘤的發生發展[5],但通過GEPIA數據庫的大樣本分析發現,與正常乳腺組織相比乳腺癌組織中MDM4的表達量無顯著變化,提示在乳腺癌發生發展過程中缺乏推動MDM4表達量變化進而參與乳腺癌的因素。進一步從生物學機制而言,SNP是通過影響基因、蛋白的表達而發揮生物學作用[6],而meta分析發現MDM4rs4245739與乳腺癌無關,由此提示MDM4rs4245739這一SNP位點可能對MDM4表達無顯著影響。此外,從統計學分析角度看,我們在納入的文獻中觀察到顯著的異質性,異質性是指納入研究結果之間的差異程度。強異質性會導致meta分析的匯總數據的評估效度降低。為了進一步探討納入研究中異質性的可能來源和其他影響異質性的積極因素,我們根據種族、地區、樣本量、基因分型檢測方法和健康對照者的來源進行了亞組分析和meta回歸分析。結果表明,健康對照者的地區分布和來源可能是異質性的主要來源。值得注意的是,這一結論是納入Montserrat等[10]的研究而得出的。考慮到這篇文章是異質性的來源,為此我們刪除了該文獻[10]并重新合并,在移除該研究后確實改變了結論,即MDM4rs4245739等位基因模型、隱性基因模型、純合子基因模型均提示其基因多態性與乳腺癌易感性之間呈負相關,這與Wang等[9]的結論一致;但在移除該研究[10]后,在顯性基因模型和雜合子基因模型中,MDM4rs4245739基因多態性仍與乳腺癌易感性無關。為此,我們試圖通過敏感性分析來探索混雜因素,結果發現Montserrat等[10]的研究對meta分析結論有影響,這可能與其樣本量大有關。而Begg’s漏斗圖和Egger’s線性回歸提示存在發表偏倚可能,由于納入研究數量少,有可能影響上述方法對發表偏倚的檢驗效能。
當然該meta分析也存在一些局限性:第一,分析的樣本量較少,有限的樣本量往往伴隨著選擇的偏差。第二,在整體的meta分析和亞組分析中都觀察到異質性,這表明潛在的因素可能會導致研究之間的異質性,我們利用隨機效應模型最小化了這一問題的可能性。第三,MDM4rs4245739位于一個約230kb的連鎖不平衡區,該區也包含tRNALys轉錄本和癌基因PIK3C2B[10],MDM4基因多態性的基因-基因相互作用對乳腺癌發生發展的影響未能闡明。第四,不同分子亞型乳腺癌具有不同的生物學、臨床和分子特征[27],但由于納入研究數據有限,無法按照分子分型進行亞組分析。
綜上所述,MDM4rs4245739基因多態性與乳腺癌易感性無相關性。考慮到上述局限性,未來需要按照乳腺癌分子分型進行大規模的病例對照研究,以進一步明確兩者之間的關系。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中老年保健(2022年6期)2022-08-19 01:41:48
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
中國生殖健康(2019年2期)2019-08-23 08:11:42
中國生殖健康(2019年6期)2019-01-06 09:20:12
電子制作(2018年18期)2018-11-14 01:48:24
祝您健康(2018年5期)2018-05-16 17:10:16
山東工業技術(2016年15期)2016-12-01 05:31:22
