基于分子對接、分子動力學模擬虛擬篩選中藥來源的醛脫氫酶1A1 抑制劑
2023-01-31 08:43:24吳輝淵鄧蘭李芬芬
中國現代藥物應用 2022年24期
關鍵詞:中藥
吳輝淵 鄧蘭 李芬芬
治療耐藥性是臨床腫瘤學面臨的最大挑戰之一[1],惡性腫瘤復發源于耐藥性腫瘤細胞能夠重新填充腫瘤生態位并播種新的病變,這些細胞被稱為癌癥干細胞(cancer stem cells,CSCs),CSCs 具有腫瘤原始特性、干細胞性和侵襲性,其重要特征之一是高醛脫氫酶1A1(aldehyde dehydrogenas 1A1,ALDH1A1) 表達[2]。ALDH1A1 主要存在于腫瘤干細胞表面及胞漿,作為CSCs 標志物,表達水平越高患者預后越差,與ALDH1A1 相關的抑制CSCs 增殖和耐藥的抗癌治療逐漸受到重視,目前尚無以ALDH1A1 為靶點的藥物上市[3]。NCT-501 對 ALDH1A1 具有較高的選擇性抑制作用,是目前ALDH1A1 活性研究中常用的陽性對照藥[4]。中藥活性成分以種類多樣、結構獨特、生物活性高等特點,在抗腫瘤領域具有突出的優勢。本研究利用類藥性篩選,分子對接,分子動力學模擬等一系列技術,從中藥化合物庫篩選出潛在的ALDH1A1 抑制劑活性物質,提供后續臨床及藥物研究的基礎。
1 材料與方法
1.1 材料 本研究采用Ledock 軟件進行分子對接工作。ADME 預測采用SwissADME 在線工具,分子動力學模擬使用AMBER 18 軟件進行。
實驗儀器與試劑:ALDH1A1 重組蛋白購于上海澤葉生物科技有限公司,血竭素(Cochinchinenin)購于成都植標化純生物技術有限公司;NCT-501 購于上海脈鉑醫藥科技有限公司,酶標儀為Spark 10 M 型,氧化型輔酶1(NAD),二甲基亞砜、乙醛均購于美國sigma,其他試劑均為國藥分析純。
1.2 靶蛋白ALDH1A1 結構預處理 虛擬篩選使用的ALDH1A1 蛋白晶體結構從PDB 數據庫[5]中下載獲得,PDB ID 為4WPN;在對接開始之前,使用對接軟件中的Lepro 模塊對受體蛋白進行處理,包括去除水分子、鹽離子以及小分子等操作。準備靶標ALDH1A1蛋白活性口袋并產生格點文件,原結晶配體坐標作為格點盒子中心。4WPN 晶體結構中蛋白與小分子抑制劑CM053 結合模式及關鍵殘基見圖1。
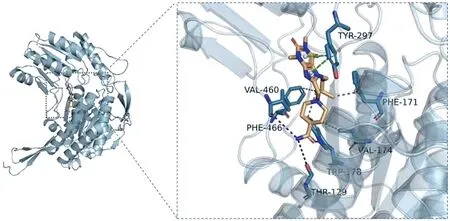
圖1 4WPN 結合模式及關鍵殘基
1.3 中藥分子化學結構的預處理 配體小分子庫含17580 個常見的天然產物分子,該數據庫從陶術生物官方(https://www.tsbiochem.com/natural-products)獲取。使用Openbabel 2.4.1 對其進行2D 至3D 格式轉換工作。
1.4 類藥性評估 基于類藥性規則“Lipinski Ro5”對中藥分子配體庫進行首輪篩選,篩選出10965 個符合類藥性規則的命中分子。Lipinski Ro5 篩選規則[6]:分子量(MW):500~700 Da,化合物的油水分配系數的對數值(clog P)=0~7.5,氫鍵供體(HBD)≤5,氫鍵受體(HBA)≤10,拓撲極性表面積(TPSA)≤200 ?2,可旋轉鍵的數量(NRB)≤20。
1.5 分子對接 設置對接盒子,使用PyMol 插件center_of_mass.py 基于活性位點的位置定義對接盒子的中心(37.33,16.60,14.42),盒子邊長設定為18 ?。設定每個分子對接輸出最大構象數為3 個,其余參數保持默認設置。輸出的打分最高的對接構象被我們認為是結合構象,選取Score<-10 kcal/mol 分子。
1.6 ADME 預測 基于ADME 藥動學參數,采用SwissADME 在線工具對Score<-10 kcal/mol 分子進行ADME 預測。從MW、HBD、HBA、脂水分配系數(lipid-water partition coefficient)、可旋轉鍵(rotatable bonds)、生物利用度(bioavailability)、TPSA、腦血管障壁(blood-Brain Barrier,BBB)、P-糖蛋白(P-gp)、水溶性ESOL Log S 等評估,篩選具有良好類藥性質的化合物。
1.7 分子動力學模擬 基于上述對接獲得的小分子與蛋白復合物作為初始結構分別進行全原子分子動力學模擬,模擬使用AMBER 18 軟件進行。模擬之前,用antechamber 模塊和gaussian 09 軟件的 HF/6-31G*計算中藥分子的電荷。之后,中藥分子采用GAFF2 力場[7],ALDH1A1 采用蛋白力場ff14SB[8]。各體系均采用LEaP 模塊給體系添加氫原子,在體系10 ? 距離處添加截斷的八面體TIP3P 溶劑盒,并在體系中添加Na+/Cl-用于平衡體系電荷,最后輸出用于模擬的拓撲和參數文件。
分子動力學模擬采用AMBER 18 軟件。在模擬之前,對體系進行能量優化,包括2500 步的共軛梯度法和2500 步的最速下降法。能量最小化后,在恒定升溫速度和固定體積條件下,在200 ps 內使體系溫度緩慢加熱到298.15 K。維持298.15 K 的體系溫度,500 ps的nvt 系綜的平衡模擬,使溶劑盒子中的溶劑分子更均勻分布。然后,對平衡后的體系在NPT 條件下模擬500 ps。最后在周期邊界性條件下分別對兩個復合體系進行100 ns 的NPT 系綜模擬。模擬時,設置10 ? 為非鍵的截斷距離,運用Particle mesh Ewald 方法計算長程的靜電作用[9],限制氫原子鍵長的方法選用SHAKE,溫控用Langevin 算法[10],其中積分步長為2 fs,體系壓強為1 atm(1 atm=1.01×105Pa),碰撞頻率γ 設為2 ps-1,每10 ps 保存軌跡數據用于進一步分析。
1.8 MM/GBSA 結合自由能計算 所有體系的蛋白和配體間的結合自由能通過MM/GBSA 方法計算[11-13]。本研究中采用45~50 ns 的分子動力學模擬軌跡用作計算,具體公式如下。
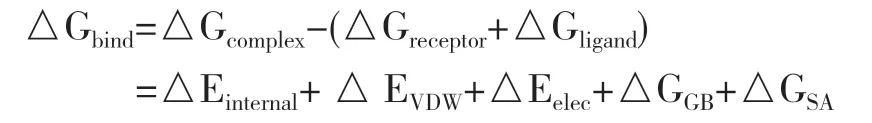
其中,△Einternal表示內能、△EVDW表示范德華作用以及△Eelec表示靜電相互作用。內能包括扭轉能(Etorsion)、角能(Eangle)、和鍵能(Ebond),△GGB和△GGA統稱溶劑化自由能。其中,GGB 為極性溶劑化自由能、GSA 為非極性溶劑化自由能。△GGB采用Nguyen[14]研究的GB 模型計算(igb=2)。△GSA 則采用表面張力(γ)與溶劑可及性表面積(surface area,SA)相乘得到,△GSA=0.0072×△SASA。
1.9 酶促反應動力學實驗 乙醛脫氫酶1 的測試方法:通過測定波長340 nm 處吸光度值每分鐘的變化,可計算出每分鐘NAD+轉化為還原型輔酶1(NADH)的量,從而得到該酶的酶活。定義每分鐘波長340 nm 處吸光度值上升1 為1 個活力單位(U)。將5 μl 濃度為150 nM ALDH1a1 加入測定孔中,反應體系在25℃,包括1 mM EDTA,2 mM NAD+,0.1 M 磷酸緩沖溶液(pH=7.4)加入測定孔,最后加入250 μM 乙醛開始反應,并開始記錄光密度(OD)值的變化,總體系反應時間為5 min,反應體系為300 μl。酶活性為△OD/min,重復試驗組3 次。
2 結果
2.1 虛擬篩選流程及結果 對接模擬技術是探究小分子與目標靶點相互作用的便捷有效手段。基于此,本研究針對ALDH1A1 蛋白的活性位點,使用ledock 軟件對常見的天然產物分子進行基于對接的虛擬篩選研究。中藥分子庫中共有17580 個中藥單體化合物,進行多輪虛擬篩選,見圖2。通過分子對接,篩選出排名前10 的命中分子,見表1。進一步ADME 預測,見表2。根據MM/GBSA 結合自由能,選取排名前4 的分子進一步做分子動力學模擬。見圖3。四個中藥化合物的名稱為血竭素(Cochinchinenin)、波棱酮(Herpetone)、2,3 二氫橡膠樹雙黃酮(2,3-Dihydroheveaflavone)、蘇鐵雙黃酮(Sotetsuflavone),對接軟件對以上化合物的結合能打分分別為-10.692、-10.309、-10.359、-10.611 kcal/mol。
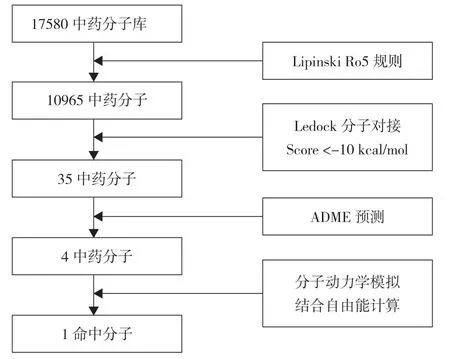
圖2 虛擬篩選流程

表1 分子對接(排名前10)
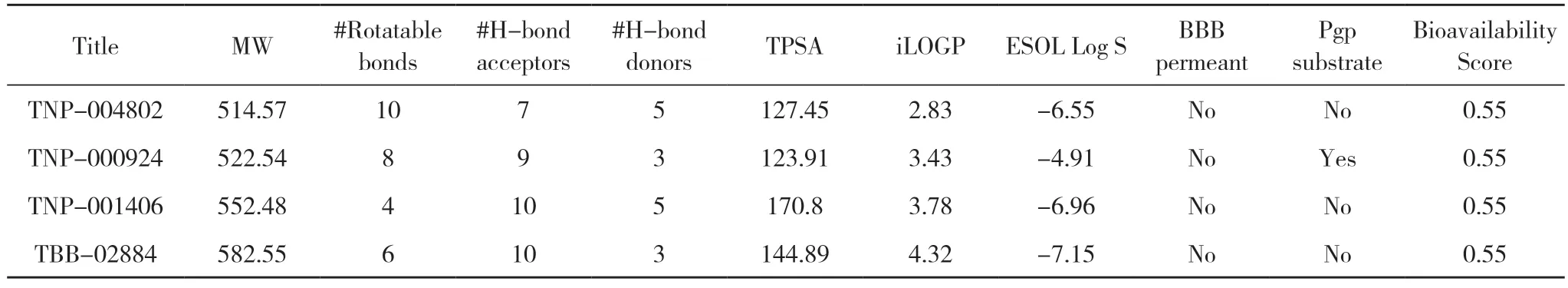
表2 ADME 參數預測(排名前4)
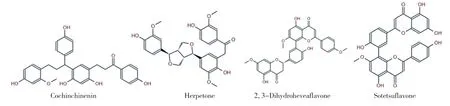
圖3 基于虛擬篩選獲得的4 個潛在的ALDH1A1 抑制分子
2.2 結合模式分析 基于上述虛擬篩選對接輸出的Cochinchinenin、Herpetone、2,3-Dihydroheveaflavone、Sotetsuflavone 與ALDH1A1 蛋白的復合物進行結合模式分析。見圖4。小分子2,3-Dihydroheveaflavone、Sotetsuflavone、Cochinchinenin、Herpetone 與ALDH1A1蛋白的結合模式:小分子2,3-Dihydroheveaflavone 與蛋白上的SER-461、TRP-178、THR-129、PHE-171、HIS-293 形成氫鍵作用,還和蛋白上的PHE-171 形成疏水作用。見圖4A。小分子Sotetsuflavone 和蛋白上的SER-461、THR-129、TRP-178、TRP-297、HIS-293形成氫鍵作用,和LYS-128、VAL-174、ILE-304 形成疏水作用。此外,Sotetsuflavone 和ALDH1A1 蛋白上的TYR-297、PHE-171 還形成pipi-共軛作用。見圖4B。對于Cochinchinenin/ALDH1A1 復合物,兩者主要通過氫鍵作用、疏水作用、pipi-共軛作用發生結合。例如小分子Cochinchinenin 和ALDH1A1 蛋白上的TRP-178、GLY-125、ASN-121、GLU-269、TYR-297 形成氫鍵作用,和ALA-462、TRP-128、LYS128、VAL-174、PHE-171、TYR-297、ILE-304、VAL-460、PHE-466 形成疏水作用。此外,小分子Cochinchinenin 還和蛋白上的PHE-171 形成pipi-共軛作用。見圖4C。對于Herpetone/ALDH1A1 復合物,小分子Herpetone 和蛋白上的TRP-178、THR-129、ASN-121 形成氫鍵作用,和VAL-174、TYR-297、PHE-290 發生疏水作用,和TYR-297、HIS-293 形成pipi-共軛作用。見圖4D。進一步,對上述四組復合物進行分子動力學模擬,來分析這些復合物的結合穩定性以及更為精確的結合能。
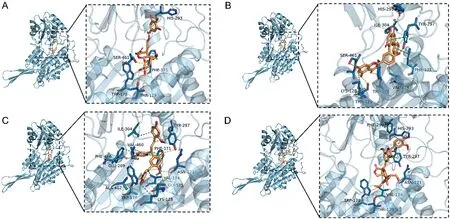
圖4 基于對接獲得的小分子2,3-Dihydroheveaflavone、Sotetsuflavone、Cochinchinenin、Herpetone 與ALDH1A1 蛋白的結合模式,左圖為整體視圖,右圖為局部視圖,圖中藍色stick 為小分子,淡青色Cartoon 為蛋白,氫鍵作用以黃色虛線顯示,疏水作用以灰色虛線顯示,pipi-共軛作用以綠色虛線顯示
2.3 穩定性分析 分子動力學模擬的均方根偏(RMSD)可以反映復合物的運動過程,RMSD 越大以及波動越劇烈表示運動劇烈,反之運動平穩。見圖5。ALDH1A1 蛋白在未結合藥物的情況下(黑線)模擬過程中波動程度大,而結合了藥物后波動程度呈現更穩定的趨勢,尤其是在ALDH1A1/TNP-004802、ALDH1A1/TNP-000924 復合物,它們在分子動力學模擬過程中RMSD 基本上在3 ? 以內穩定波動。暗示ALDH1A1/TNP-004802、ALDH1A1/TNP-000924 復合物結合非常穩定。
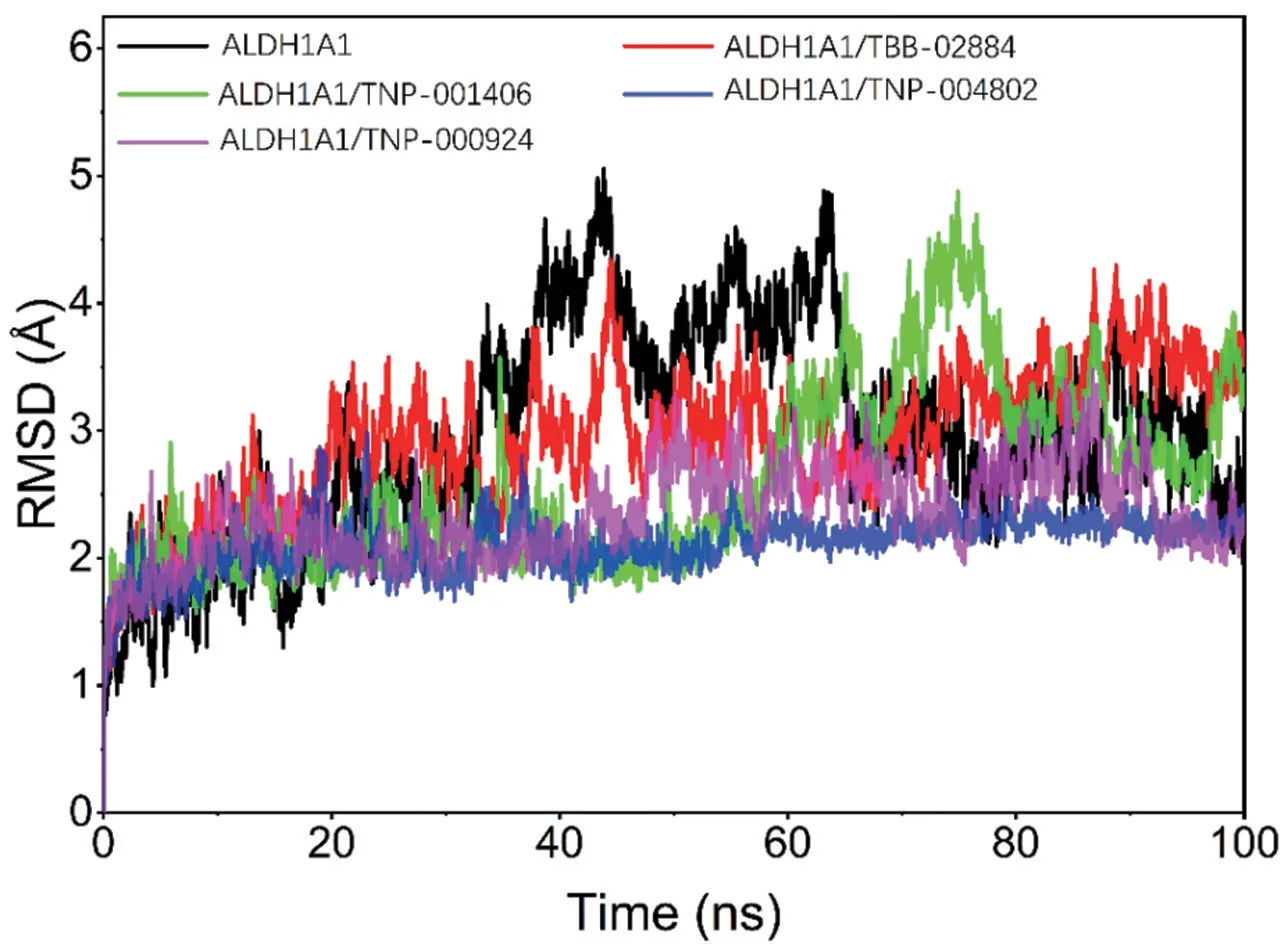
圖5 分子動力學模擬過程中復合物RMSD 差隨時間的變化
均方根波動(RMSF)可以反映分子動力學模擬的過程中蛋白的柔性。通常藥物結合蛋白后,蛋白柔性下降,進而達到穩定蛋白的作用,發揮酶活效果。除了蛋白兩端以及130~145 段氨基酸外,其余各個序列段的RMSF 數值偏低,表示整個結構的柔性較低。對比結合化合物和未結合化合物蛋白的RMSF 數值,發現結合化合物時,蛋白的RMSF 更低,其中結合TNP-004802最顯著,其次為TBB-02884、TNP-000924、TNP-001406。見圖6。
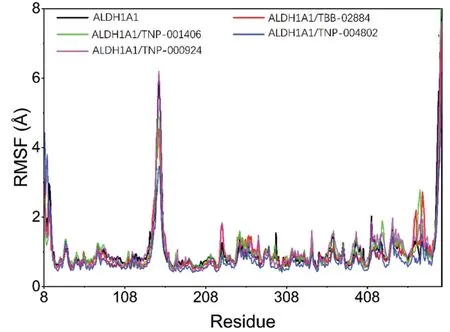
圖6 基于分子動力學模擬軌跡計算的RMSF
2.4 MM/GBSA 結果 在本研究中基于MM/GBSA 計算方法,對分子動力學模擬軌跡充分采樣以準確計算小分子與蛋白的結合能。ALDH1A1 蛋白和TBB-02884、TNP-001406、TNP-004802、TNP-000924 的結合能分別為(-36.35±3.70)、(-43.61±2.32)、(-56.66±3.83)、(-52.26±2.64) kcal/mol。四個組合的結合能都非常高,其中ALDH1A1 蛋白和TNP-004802、TNP-000924 的結合能甚至>-50.0 kcal/mol,暗示其結合較好。通過殘基分解,四個組合結合的主要貢獻力為疏水作用,其次是靜電作用,最后是非極性溶劑化自由能。見表3。
表3 MM/GBSA 計算結合自由能(,kcal/mol)
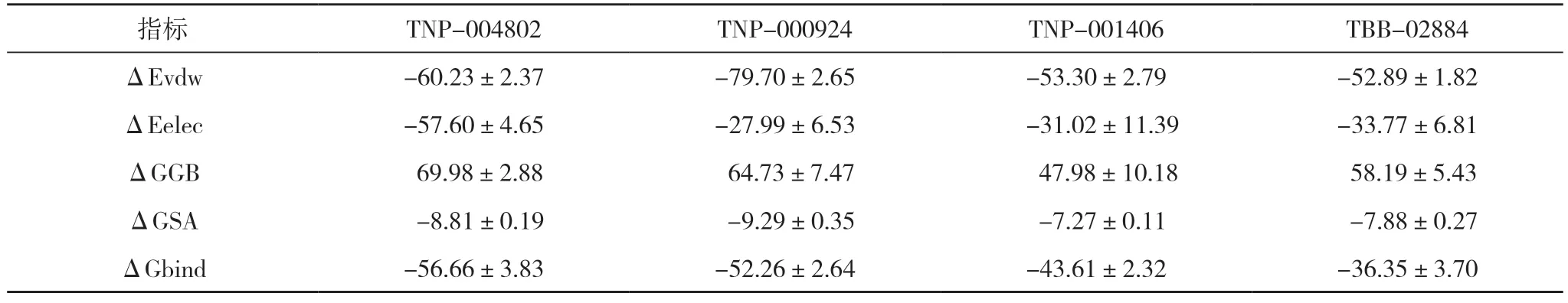
表3 MM/GBSA 計算結合自由能(,kcal/mol)
ΔEvdW: 范德瓦耳斯能量(van der Waals energy);ΔEelec: 靜電能量(electrostatic energy);ΔGGB: 靜電對溶劑化的貢獻(electrostatic contribution to solvation);ΔGSA: 溶劑化的非極性貢獻(non-polar contribution to solvation);ΔGbind: 結合自由能(binding free energy)
ALDH1A1 上的氨基酸類型對該結合起重要作用,對理解結合機制也非常重要,將4 個候選分子與ALDH1A1 結合的能量分解至每個殘基的貢獻。復合物中對結合能起貢獻的Top-10 氨基酸:對于ALDH1A1/TBB-02884 復合物,ILE 304、PHE 289、PHE 170、ASN 121、TRP 178、TYR 297、VAL 460、TYR 457、CYS 302、VAL 459 為關鍵性氨基酸;對于ALDH1A1/TNP-001406 復合物,TRP 178、TYR 297、VAL 460、ASN 121、PHE 171、ILE 304、SER 461、GLY 125、LYS 128、ALA 462 為結合貢獻大的氨基酸;對于ALDH1A1/TNP-004802 復合物,TRP 178、ILE 304、VAL 174、TYR 457、VAL 460、PHE 171、TYR 297、CYS 302、ASN 121、SER 461 為結合貢獻大的氨基酸;對于TNP-000924 復合物,TYR 297、GLY 125、ALA 462、VAL 460、TRP 178、PHE 290、VAL 459、SER 461、GLN 463、VAL 174 為結合貢獻大的氨基酸。見圖7。
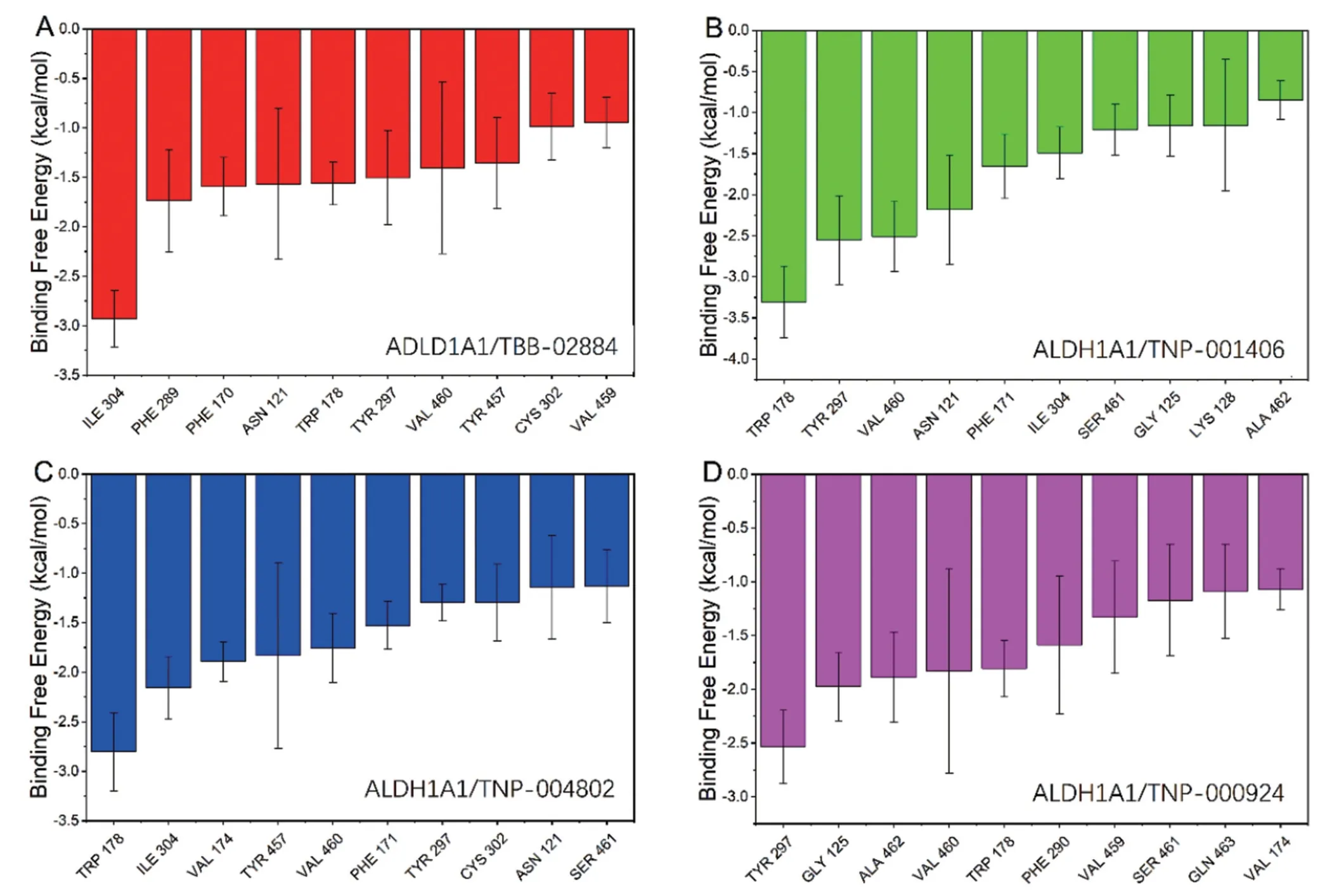
圖7 對小分子和蛋白結合能起貢獻的Top-10 氨基酸
2.5 氫鍵分析 氫鍵作用是小分子化合物和蛋白結合過程中最強的非共價作用之一,在分子動力學模擬期間,統計各個復合物形成的氫鍵情況:小分子TBB-02884、TNP-001406、TNP-004802、TNP-000924 和ALDH1A1 蛋白在模擬過程中形成的氫鍵數目都維持在2 個以上。表明氫鍵作用對四個復合物穩定存在起重要作用。TNP-001406、TNP-000924 在模擬過程中和ALDH1A1 蛋白形成的氫鍵頻率要高于TBB-02884、TNP-004802,意味著TNP-001406、TNP-000924 和ALDH1A1 蛋白的結合更加依賴于氫鍵作用。見圖8。
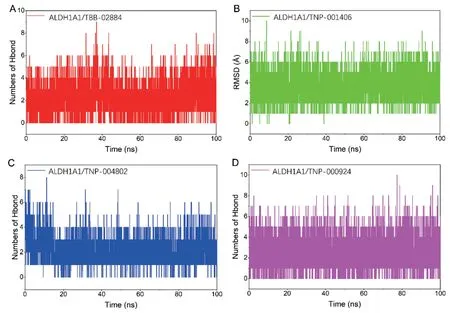
圖8 分子動力模擬過程中小分子與蛋白的氫鍵數目變化
2.6 酶活抑制實驗結果 TNP-004802 與ALDH1A1結合穩定,且結合能最高,進一步運用酶動力學實驗方法比較TNP-004802 與NCT-501 對ALDH1A1 的抑制活性,結果顯示,兩者均有較好的抑制活性,低濃度的TNP-004802 抑制活性高于NCT-501。見圖9。
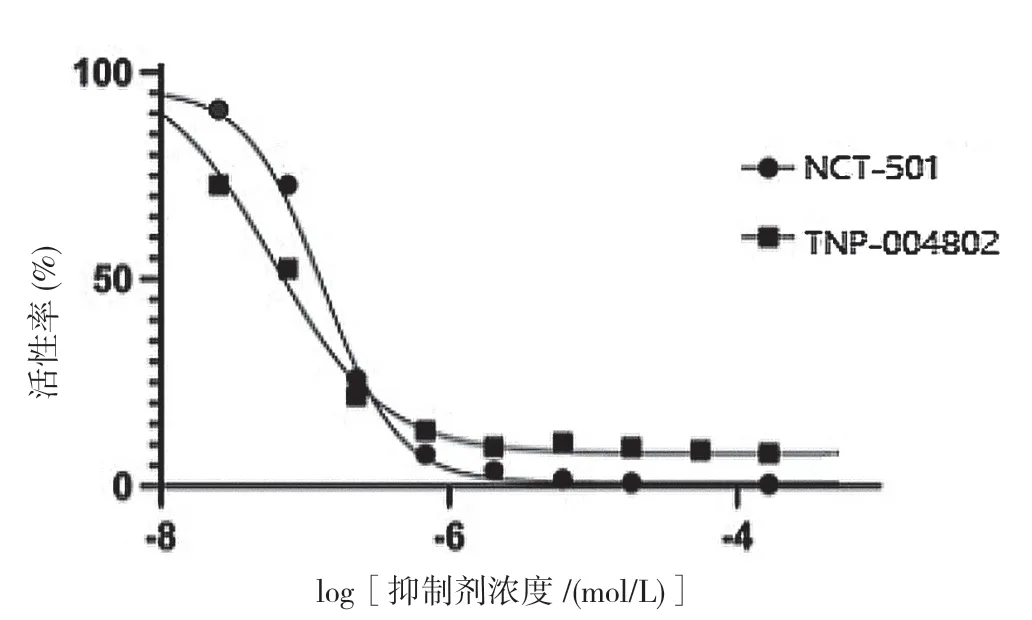
圖9 TNP-004802 與NCT-501 對ALDH1A1 的抑制活性
3 討論
醛脫氫酶(ALDHs)是一類依賴于NAD(P)的酶,可催化醛氧化成羧酸。ALDHs 是四聚體酶,N 端的結合域與輔因子 NAD+結合,從而改變四聚體構象[15]。催化位點的亞基結構形成結合口袋與乙醛等底物結合后發生結構改變,這種相對柔性的結構改變是限制醛氧化的關鍵步驟[16]。人體中,已知有19 種功能性基因編碼ALDHs 的基因[17]。視黃醛的主要催化酶是ALDH1A1。目前分離到的多種CSCs 發現ALDH1A1表達水平升高[18],聯合CD133、CXCR4、Notch-4 等標志物用于分離和鑒定CSCs。因此,ALDH1A1 可作為抗腫瘤治療的重要靶標,研發 ALDH1A1 抑制劑是目前腫瘤治療領域的熱點,目前已發現多種不同結構的抑制劑,未來研究方向將主要在先導化合物的優化。許多腫瘤細胞系以及患者中ALDH1A1 酶水平升高導致對化療和放療的耐藥[19]。
ALDHs 抑制劑雙硫侖適合從腫瘤中根除化學抗性干細胞樣腫瘤起始細胞和預防復發的治療方法[20]。Omran[21]開發了雙硫侖衍生物2b 顯示出與雙硫侖對ALDH1A1 相當的活性。Verma 等[22]通過三維定量構效關系和骨架躍遷設計了21 種ALDH1A1 抑制劑,全部化合物均顯示半抑制濃度(IC50)值在0.02~0.80 μM的范圍內,表明這些作為環磷酰胺耐藥的輔助治療的潛力。研究表明[23],乙醛脫氫酶1 的催化活性中心主要為半胱氨酸-302(Cys302),已報道的抑制劑主要通過與Cys302 結合導致酶的催化活性受抑制。本研究對17580 個中藥分子進行Lipinski Ro5 規則、分子對接、ADME 預測,選取了4 個化合物進行分子動力學模擬,Cochinchinenin、Herpetone、2,3-Dihydroheveaflavone、Sotetsuflavone。本結果發現Cochinchinenin 與ALDH1A1的Cys302、TRP 178 等多個氨基酸殘基位點結合,穩定性最顯著,結合能最高,酶活抑制實驗顯示了其對ALDH1A1 良好的抑制活性。
Cochinchinenin 是來源于中藥血竭的化合物,《唐本草》論及血竭的功效為“主五臟邪氣,帶下,止痛,破積血,金創生肉”,常用于跌打損傷、惡瘡、瘰疬等。血竭一直以來用于惡性腫瘤的治療,明代龔廷賢所著《萬病回春·積聚》提及含血竭的藥方”神化丹”之功效:“消癖積、破血塊、下鬼胎、通經脈及諸痞積血氣塊”。現代臨床應用單味血竭粉外敷、內服治療癌性潰瘍108 例,取得滿意療效[24],外敷可用于癌性疼痛[25],實驗研究方面,血竭素高氯酸鹽通過降低細胞線粒體膜電位,誘導人乳腺癌細胞MCF-7 凋亡[26];通過活化MAPK 通路中的JNK 和p38 發揮促進膠質瘤細胞LU251 凋亡的作用[27]。
本研究虛擬篩選血竭素作為ALDH1A1 抑制劑,具有抑制腫瘤干細胞增殖和耐藥的可能藥效,血竭素可作為先導化合物研究,進一步可開展藥物化學、藥理學及臨床研究,找到與ALDH1A1 相關的抑制癌癥干細胞增殖和耐藥的有效藥物。
猜你喜歡
中老年保健(2021年5期)2021-12-02 15:48:21
中老年保健(2021年4期)2021-12-01 11:19:40
中老年保健(2021年4期)2021-08-22 07:08:32
中國現代中藥(2020年10期)2020-12-16 08:53:18
金橋(2020年7期)2020-08-13 03:07:00
基層中醫藥(2020年12期)2020-07-22 06:34:38
中國現代中藥(2020年4期)2020-06-10 09:56:34
基層中醫藥(2018年6期)2018-08-29 01:20:20
長春中醫藥大學學報(2017年1期)2017-04-16 05:56:49
肝博士(2015年2期)2015-02-27 10:49:49
