新生兒糖尿病6例病例特點及臨床分析
2023-02-01 12:45:26王淑琴高怡青朱磊侯夢劉金鳳薛穎
安徽醫藥 2023年2期
王淑琴,高怡青,朱磊,侯夢,劉金鳳,薛穎
新生兒糖尿病(neonatal diabetes mellitus,NDM)多在6月內發病,少數可發生于1歲前,病因以單基因突變為主[1]。隨著對NDM認識的提高,發病率上升,最近研究顯示其發病率為 1/9 000~1/160 000[2]。NDM按病程分為永久性新生兒糖尿病(permanent neonatal diabetes mellitus,PNDM)和暫時性新生兒糖尿病(transient neonatal diabetes mellitus,TNDM),以NDM發病的綜合征一般歸類為PNDM。PNDM可伴有部分或完全的胰島素缺乏,診斷時臨床表現包括宮內生長遲緩、高血糖、糖尿、滲透性多尿、糖尿病酮癥(diabetic ketosis,DK)、糖尿病酮癥酸中毒(diabetic ketoacidosis,DKA)等,目前已明確的基因突變包括ABCC8、GCK、INS、KCNJ11、PDX1等,病程因基因型不同而各不相同[3]。TNDM一般在發病后18個月內緩解,染色體6q24差異性甲基化異常是TNDM常見病因[4],約半數以上的TNDM在兒童期或青春期復發,復發后可予磺脲類藥物或較小劑量的胰島素治療,優選治療方案仍需進一步探討[5]。TNDM診斷時較少發生DKA,所需胰島素治療劑量低。此外,以NDM發病的綜合征如Wolcott-Rallison綜合征、IPEX綜合征、Wolfram綜合征等,對糖尿病治療仍以胰島素治療為主,伴發癥狀不同預后亦不同。對于新生兒期起病的NDM很難直接診斷PNDM、TNDM,在隨訪過程中根據治療情況而定。明確基因的致病性變異對于臨床治療重要,多數攜帶KCNJ11或ABCC8致病性變異的病人可使用磺脲類藥物治療。使用磺脲類藥物治療不僅可以改善血糖控制,亦可改善病兒的神經認知功能[6-7]。本研究通過對6例NDM臨床特點及隨訪情況進行分析,為臨床診療提供依據。
1 資料與方法
1.1 一般資料選擇2013年2月至2021年6月于徐州醫科大學附屬徐州兒童醫院就診的6例NDM為研究對象,收集臨床資料進行分析。本研究已獲得徐州醫科大學附屬徐州兒童醫院醫學倫理委員會批準(倫2021-06-01-K05)。
1.1.1 診斷標準 6月內嬰兒持續高血糖(血漿葡萄糖濃度150~200 mg/dL),同時排除應激、感染、藥物等引起的一過性高血糖,對于1歲內發病經基因檢測證實亦可診斷為NDM。
1.1.2 診斷和治療方法 收集6例NDM病兒臨床資料,包括:出生史、出生體質量、性別、C肽、糖化血紅蛋白(HbA1C)、抗胰島細胞抗體(ICA)、抗胰島素抗體(IAA)、抗谷氨酸脫羧酶抗體(GADA)、靜脈血葡萄糖、血氣分析、血電解質及尿酮等。6例病兒均住院治療。合并糖尿病酮癥酸中毒(DKA)或糖尿病酮癥(DK)者采用補液及小劑量胰島素糾酮體治療。DKA、DK糾正后采用胰島素皮下注射方案。5例病兒經用格列本脲口服替代治療,用量從0.1 mg·kg-1·d-1, 1至 2 周內逐漸增加至 0.8 mg·kg-1·d-1,同時逐漸減少胰島素用量直至停用胰島素。如停胰島素后,格列本脲治療可平穩控制血糖,提示治療有效。如格列本脲增至0.8 mg·kg-1·d-1,仍不能減少胰島素用量,提示格列本脲治療無效。
C肽測定采用化學發光法(瑞士羅氏公司),ICA、IAA、GADA測定采用酶聯免疫吸附試驗(瑞士羅氏公司),HbA1C測定采用高效液相色譜法。
基因檢測方法:病兒近親屬知情同意后,根據徐州醫科大學附屬兒童醫院基因檢測相關流程,抽取血標本(病人及父母靜脈血各 2 mL)送至第三方進行檢測。病例 1、2、3、4、6均送至南京金域醫學檢驗,病例5送至康圣環球海斯特。南京金域醫學檢驗公司通過Ion-Torrent-PGM技術對新生兒糖尿病相關基因進行測序。康圣環球海斯特使用對應試劑盒提取樣本 DNA 后,使用探針(GenCap)捕獲相關候選基因進行高通量測序。
2 結果
2.1 一般資料6例NDM男孩5例,女孩1例。合并小于胎齡兒3例,病兒診斷年齡為3~256 d,中位年齡為50 d。隨訪時間4.5~105.2個月。1例因病兒診斷NDM后發現其母親為空腹血糖受損,余病例無糖尿病家族史。見表1。
2.2 初次診斷時情況3例以發熱感染為首發癥狀,3例以反應差起病。初診時4例合并糖尿病酮癥酸中毒,1例合并糖尿病酮癥。診斷時平均糖化血紅蛋白 11.4%(7.35%~13.8%),C肽均降低,ICA、IAA、GADA抗體均陰性。急性期均用胰島素治療,血糖控制良好。5例病兒行格列本脲經驗性治療,4例有效,1例無效停藥,成功率為80%,均無明顯不良副作用。見表1。
2.3 隨訪情況隨訪時間最短4.5月,最長8年6月。1例TNDM,4例PNDM,1例隨訪4.5月并發肝功能衰竭死亡,因隨訪時間短未分型。6例行基因檢測,KCNJ11基因突變4例,其中1例為首次報道。1例TNDM急性期予胰島素治療后,轉為格列本脲治療成功,病兒6月齡時停格列本脲,8歲時糖尿病復發,予長效小劑量胰島素治療血糖可控制平穩。3例使用格列本脲PNDM隨訪至今血糖均控制良好,無明顯不良反應,經基因檢測證實均為KCNJ11突變所致。1例使用胰島素治療PNDM,隨訪至今1歲4月,血糖波動相對較大。5例生長發育與同性別同齡兒相仿,無語言運動發育落后。見表1。
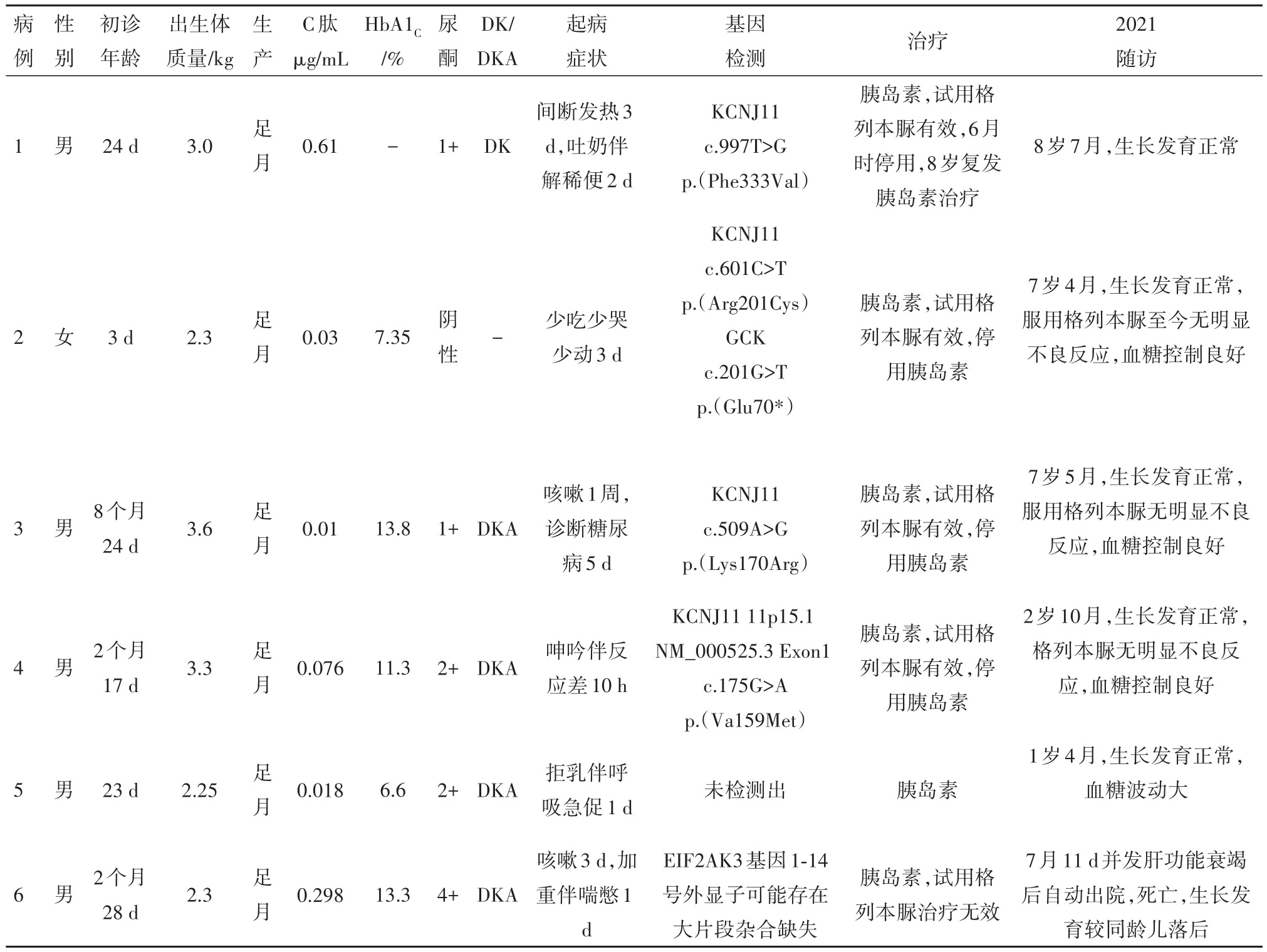
表1 6例新生兒糖尿病臨床特點分析
3 討論
新生兒糖尿病多指小于6個月嬰兒的持續性高血糖(血漿葡萄糖濃度 150~200 mg/dL)。然而最近的研究發現,單基因形式的NDM在1歲時仍可發病[5,8],因此推薦1歲以下發病的糖尿病病人行基因檢測以精準化指導診療[9]。單基因NDM的預后和治療方案在很大程度上取決于哪些基因受到影響。基因檢測使NDM精準化治療比例得到很大提高[10]。正如本文中病例3,初診年齡8月余,發病年齡超過傳統關于NDM認知6月齡內,行基因檢測明確為KCNJ11突變,行格列本脲治療有效,停用胰島素,服用格列本脲控制血糖良好,無明顯低血糖事件發生,已隨訪至7歲5月,病兒生長發育正常,此例病兒精準化治療得益于基因檢測。
TNDM的兒童通常在嬰兒期(13~18周齡)高血糖得到緩解。然而,在青春期或成年期可能會復發[11]。TNDM最常見的原因6q24基因座上的基因過度表達,由于6號染色體單親二倍體、該區域的異常復制(父系復制)或DNA甲基化的丟失,從而激活母體等位基因而導致6q24處印記缺失的結果[12]。第二個最常見原因是編碼電壓依賴性鉀通道亞單位(KATP)的兩個基因突變[13],分別為KCNJ11編碼的Kir6.2亞單位,ABCC8編碼SUR1亞單位[14],這兩個基因中的任何一個激活突變都會導致KATP通道不適當地開放,從而導致盡管高血糖但細胞膜無法去極化和胰島素釋放障礙。
本研究中病例1為TNDM病兒,以發熱感染就診,合并糖尿病酮癥,胰島素治療病情穩定后,轉換為格列本脲并停用胰島素治療,血糖控制良好。6月大時病兒病情緩解,停用格列本脲,本例病兒緩解期較文獻報道晚,但在隨訪過程至8歲時糖尿病復發,調整為小劑量胰島素治療。此例病兒基因檢測為KCNJ11雜合突變,Phe333Val突變未見任何文獻報道,生物信息學認為其對KCNJ11基因功能存在一定影響,結合父母均未檢出Phe333Val突變,結合診療隨訪情況,其突變考慮極可能為新發突變位點導致TNDM。
PNDM最常見的原因是激活KCNJ11或ABCC8的雜合突變[15-16],占所有新生兒糖尿病病例50%以上。這兩種基因突變也是TNDM的第二位常見原因。這兩個基因突變的病人對磺脲類藥物治療敏感[17]。大約25%的ABCC8或KCNJ11基因突變病人患有神經認知功能障礙,從精神運動障礙到與嚴重癲癇相關的認知發育遲緩[18],使用磺胺類藥物治療使HbA1C正常化,同時減少ABCC8或KCNJ11突變的新生兒糖尿病中低血糖的發生率,亦可改善神經認知功能[19]。繼KCNJ11、ABCC8基因突變之后,第二種最常見的永久性新生兒糖尿病是胰島素基因INS突變。INS突變可出現在20%的PNDM嬰兒中。該基因突變導致胰島素蛋白錯誤折疊,從而導致內質網應激增加和最終β細胞死亡[20]。 INS突變病人的診斷時間晚于ATP敏感性鉀離子通道突變攜帶者(11周比 8周)[8]。除永久性新生兒糖尿病外,病人沒有任何其他表型特征,需要終身胰島素治療[21]。
本研究中1例經基因檢測未發現明確致病基因的PNDM,故未予格列本脲轉換治療,診斷后予胰島素治療至今,生長發育正常,但血糖控制因年齡小波動相對較大。另3例予格列本脲治療有效PNDM病兒,經基因檢測證實均為KCNJ11基因突變。此3例病兒隨訪至今,生長發育正常,目前關于格列本脲長期服用相關療效及副作用報道極少,本組病例中最長已服用7年4個月,可平穩控制血糖,且無明顯不良反應,提示格列本脲相對較安全可靠。
KCNJ11基因突變3例病兒中有1例合并GCK雜合突變,該病兒因“少吃少哭少動3 d”入院就診,入院后結合基因檢測KCNJ11基因突變診斷為新生兒糖尿病,格列本脲治療有效,此外該病兒合并GCK基因雜合突變,可導致MODY2發生,基因檢測證實GCK基因突變來源母親,后母親監測血糖發現空腹血糖高,無臨床癥狀,目前經飲食干預及生活鍛煉后糖化血紅蛋白可維持正常范圍,提示該病兒未來亦可能合并MODY2發生,目前仍需繼續隨訪該病兒病情進展情況。
KCNJ11突變病人可能存在廣泛的神經認知障礙,睡眠障礙,注意缺陷多動障礙以及行為發育延遲[22]。本研究中3例病兒經診斷后及時予格列本脲治療,隨訪至今未發現嚴重低血糖事件,生長發育與同性別同齡兒相仿,提示格列本脲可平穩控制KCNJ11突變導致NDM病兒的血糖,同時可能對病兒神經認知發育有改善作用,但本研究長期使用格列本脲治療KCNJ11突變導致的NDM僅3例,未來仍需更多相關的病例納入研究。這也強調對于KCNJ11突變導致的NDM病兒中,格列本脲比胰島素治療不僅更好的控制病兒血糖,并可改善神經認知功能,建議NDM診斷后盡早行基因檢測以提供精準化治療。
NDM還包括與NDM相關的綜合征,一般歸類于PNDM,發病機制包括胰島β細胞破壞、胰腺發育不全或再生障礙、胰島β細胞功能受損或嚴重胰島素抵抗等[1]。最常見的綜合征是Wolcott-Rallison綜合征(Wolcott-Rallison synkdrome,WRS),是一種由EIF2A突變引起的常染色體隱性疾病,EIF2A是一種編碼翻譯起始因子2-α激酶3的基因,在內質網的調節中起重要作用。其他特征是肝功能障礙和骨骼發育不良[23]。
任何患有與骨骼發育不良和、或急性肝衰竭有關的PNDM,都應懷疑患有 WRS[24]。由于該綜合征的主要表現特征可能發生在疾病過程的后期,因此,在任何新生兒或6個月之前發生糖尿病的嬰兒中,應懷疑患有WRS。其臨床過程非常多變,糖尿病發病初期,骨發育不良和反復肝衰竭是病人最典型的特征[25]。通常,糖尿病發生在6個月之前,骨發育不良在一兩歲之前。肝衰竭可能發生在疾病過程中的任何時間,并可能在糖尿病發病后首次出現,由于病人年齡小,一些臨床表現可能缺失,在進行 WRS 的完整診斷之前可能會發生死亡。本研究中病例6,2個月28 d時以“咳嗽3 d,加重伴喘憋1 d”入院,入院后完善檢查示糖尿病酮癥酸中毒,予胰島素治療病情穩定后,試用格列本脲治療無效,基因檢測示EIFIA3基因1~14號外顯子可能存在大片段雜合缺失。隨訪中病兒合并3次肺部感染,7個月11 d時并發肝功能衰竭后自動出院死亡,此例病兒先發生NDM,隨后出現肝功能衰竭,未發現骨發育不良,考慮病程短,仍應考慮WRS可能。
綜上所述,1歲內發病糖尿病病人應考慮基因檢測,因為NDM的臨床表型與基因型異質性密切相關。磺胺類藥物可以用于治療 KATP通道基因突變(KCNJ11和ABCC8),并可改善神經認知功能。建議所有新生兒糖尿病病人進行密切隨訪,并關注磺脲類藥物長期治療反應。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國生殖健康(2020年2期)2021-01-18 02:51:26
家庭醫學(下半月)(2019年9期)2019-10-12 08:04:06
家庭醫學(下半月)(2019年8期)2019-09-25 09:02:00
中國生殖健康(2019年2期)2019-08-23 08:12:10
小學生導刊(2018年13期)2018-06-29 03:49:00
媽媽寶寶(2017年3期)2017-02-21 01:22:12
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
中國衛生標準管理(2015年8期)2015-01-26 18:08:35
