田螺科4種代表性貝類基因組調研及SSR特征分析
2023-05-08 06:23:22季秋婷王俊杰唐伯平
江蘇農業科學 2023年6期
關鍵詞:物種
朱 穎, 陳 瑜, 唐 俊, 季秋婷, 王俊杰, 唐伯平, 王 剛
(鹽城師范學院濕地學院/江蘇省鹽土生物資源研究重點實驗室,江蘇鹽城 224007)
田螺科(Viviparidae)貝類是我國淡水水域常見的軟體動物,其屬于腹足綱、中腹足目,廣泛分布于湖泊、沼澤、水庫、池塘及溪流等處[1],能對水體健康起到維持作用,凈化水體,是重要的生態指示物種[2]。田螺科物種多為經濟貝類,其營養價值高,螺肉鮮美,是低脂肪、高蛋白食品,在我國廣受歡迎,同時也是家禽,水產養殖魚類、蝦蟹類的天然飼料[3]。田螺科貝類屬于世界物種,我國包含9個屬,70多個種,其中河螺屬(Rivularia)和螺螄屬(Margarya)為特有屬,環棱螺屬和圓田螺屬物種最為常見[4]。隨著資源環境的破壞和對田螺的需求日益增加,加上外來淡水螺的不斷入侵,我國本土田螺科物種分布和數量受到嚴重威脅,很多種群在數量上明顯衰退,嚴重的已瀕臨滅絕[5]。基于田螺科物種的重要生態價值和經濟價值,保護和恢復田螺科物種的多樣性及種群數量的問題顯得尤為重要。
隨著組學時代的到來和發展,測序成本下降,高通量測序被廣泛用于軟體動物基因組測序中。在軟體動物中,首個貝類——太平洋牡蠣(Pacific oyster)基因組圖譜于2012年完成,標志著軟體動物組學時代的到來,填補了軟體動物家族基因組圖譜的空白[6]。隨后,霸王蓮花青螺(Lottiagigantea)[7]、章魚(Octopusbimaculoides)[8]、牡蠣(Crassostreavirginica)[9]等其他軟體動物的基因組圖譜陸續發表,這為其他軟體動物的基因組圖譜的構建提供了基礎。截至2022年6月,在NCBI數據庫中已公布92個軟體動物基因組圖譜。基因組的解析可為貝類優良性狀基因挖掘、遺傳機制解析提供研究基礎。然而,目前淡水的田螺科貝類分子生物學研究基礎相對薄弱,關于田螺科的研究大多集中在初步調查、系統發育和分類地位等方面的分析[10-12]。在NCBI數據庫中,提供的田螺科分子數據十分有限,缺乏田螺科貝類參考基因組信息。在構建基因組精細譜圖之前進行基因組特征調研,可為后續的測序方案提供參考依據。目前,主要采用K-mer分析方法對基因組進行調研評估獲取物種雜合率、重復度等信息[13-15]。本研究選取淡水田螺科的中華園田螺(Cipangopaludinacathayensis)、銅銹環棱螺(Bellamyaaeruginosa)、多棱角螺(Angulyagrapolyzonata)、湄公螺(Mekongiarivularia)等4種代表性貝類進行基因組調研,通過K-mer分析,進行物種雜合率、基因組重復序列比例及基因組大小的評估,以期為田螺科貝類基因組學研究提供參考。同時,利用Illumina 測序數據對4個田螺科貝類進行初步組裝和初步比較分析,采用MISA工具對簡單重復序列(SSR)進行鑒定和分析,為進一步分子標記開發和資源鑒定提供研究基礎。
1 材料與方法
1.1 材料
中華園田螺、銅銹環棱螺、多棱角螺和湄公螺,于2021年5—6月分別采集自四川(30°23′36.82″N,104°4′50.46″E)、江蘇鹽城(33°22′33.4″N,120°12′7.19″E)、湖南(27°49′36.25″N,113°7′59.75″E)、江西(28°30′7.34″N,115°48′22.93″E),采集的田螺科樣本保存于江蘇鹽城師范學院沿海灘涂重點實驗室。
1.2 樣品DNA提取
在江蘇省鹽城師范學院沿海灘涂重點實驗室,使用Omega公司的試劑盒D3373-01 Mollusc DNA Kit(50)提取田螺科4種代表性螺的基因組DNA(取螺的部分樣品),用NanoDrop 光譜儀測定DNA 的濃度和純度。DNA樣本標準為:DNA總量≥20 μg 且濃度≥300 ng/μL;樣本純度要求為:D260 nm/280 nm≥1.8,D260 nm/230 nm≥1.8。
1.3 4種貝類樣本測序和基因組評估
DNA樣本送到武漢菲沙基因信息有限公司進行文庫制備,基于高通量測序儀(MGI-SEQ2000)平臺采取雙端150 bp(PE150)技術進行測序,測得的數據經過質控后用來進行下一步數據分析。4種貝類Illumina測序數據存儲在國家基因組科學數據中心數據庫(https://ngdc.cncb.ac.cn/),項目編號:PRJCA013294。
文庫構建參照Illumina標準規程[16],構建并插入片段大小為270 bp的文庫[17]并測序。完成構建的文庫在Illumina HiSeq 2500平臺進行測序。選用AdapterRemoval(version 2.1.7)軟件對測序數據去接頭,用FastQC軟件進行質量評估,并過濾質粒污染。采用基于K-mer分析進行估算,K值取21,在測序reads均勻分布的前提下,計算基因組大小(基因組大小=總堿基數/平均測序深度=總K-mer數平均值/平均K-mer深度)。利用Jellyfish和 GenomeScope 軟件對K-mer頻數分布數據進行統計并擬合作圖,得到K-mer分布圖。
1.4 基因組組裝和GC深度分析
對過濾數據利用SOAPdenovo軟件進行基因組組裝,將讀長(reads)分成100 bp長度的K-mer,再基于 K-mer 數據構建de Bruijn圖。利用perl腳本對組裝的基因組進行滑窗統計,設置窗口為10 kb,根據GC分布和覆蓋深度統計結果,應用R腳本繪制散點圖。
1.5 簡單重復序列(SSR)特征分析
對初步完成組裝的基因組進行簡單重復序列分析。使用微衛星識別工具(microsatellite identification tool,MISA)搜索SSR位點對于150 bp以上的序列片段(Scaffolds)[18]。同時運行MISA軟件程序的配置文件misa.ini,使用MISA默認參數。
2 結果與分析
2.1 田螺科4種貝類基因組測序質量和基因組大小評估
對Illumina Hiseq PE測序平臺測得的中華園田螺、銅銹環棱螺、多棱角螺、湄公螺等4個代表性貝類的原始數據,經過濾和校正等質控步驟后,分別獲得52.59、58.64、55.71、54.90 G的有效數據,有效數據均大于96%(表1)。測序質量評估結果顯示,4種螺的Q20分別為 96.5%、96.1%、96.4%、96.6%,Q30分別為88.5%、87.5%、88.3%、88.8%,Q20均大于95%,Q30均大于85%,表明4種貝類的測序質量較好,滿足分析要求。4種田螺科貝類GC 含量分別為33.4%、33.7%、33.1%、33.8%,GC含量基本持平。

表1 田螺科4種螺基因組調查測序結果統計
本研究采用K-mer 的分析方法,取K為21來進行物種基因組特征分析,K-mer分布圖如圖1,實際觀測的K-mer分布為藍色區域;測序頻數較低的K-mer為紅色線條下方區域(測序錯誤);黑色線條下方是可靠的K-mer數據,用于4種貝類基因組大小的評估;黃色線條下方區域是非重復區域。由圖1可知,中華園田螺、銅銹環棱螺、多棱角螺、湄公螺的主峰分別在測序深度18×、18×、45×、23×左右,均只有1個主峰,說明4種螺均為2倍體。在測序數據中去除深度異常的K-mer后,評估得到4種螺的基因組大小分別為1 353、896、991、1 196 Mb;雜合度分別為0.77%、1.37%、2.41%、0.91%。4種貝類雜合率均大于0.5%,為高雜合基因組。4種螺的重復序列比例分別為26.2%、26.7%、27.8%、28.8%,比例基本一致(表2)。


表2 田螺科4種螺基因組評估結果
2.2 田螺科4種貝類基因組序列初步組裝
利用SOAPdenovo軟件分別對4種貝類經過質控處理后得到的高質量read,進行初步組裝,中華園田螺、銅銹環棱螺、多棱角螺、湄公螺的基因組大小分別為 1 598、1 845、1 202、1 322 Mb,其中銅銹環棱螺基因組長度最大,多棱角螺基因組長度最小。4種貝類的Contigs N50分別為690、200、1 025、889 bp(表3)。初步組裝得到的基因組大小與K-mer評估得到基因組大小有差異,其組裝的基因組大小偏大,這可能雜合度有關,也有可能與二代測序數據讀長較短和覆蓋度較低等因素有關。
2.3 田螺科4種貝類初步組裝的GC含量與覆蓋度分布
對4種田螺科貝類初步組裝得到的基因組,過濾覆蓋深度10×以下的contig序列,基于覆蓋深度和GC含量繪制成散點圖。由圖2可知,中華園田螺、銅銹環棱螺、多棱角螺、湄公螺4種螺的GC含量較為一致,在27%~43%范圍內,但區域測序深度分布有所差異。其中中華園田螺測序深度較高,在20~40×區域深度分布比較集中,其次分別是湄公螺(18~25×)、多棱角螺(10~38×)、銅銹環棱螺(10~20×)。4種田螺科貝類均為1個主峰,說明無外源污染。

2.4 田螺科4種貝類SSR特征分析
對中華園田螺、銅銹環棱螺、多棱角螺、湄公螺等4種貝類初步組裝的基因組,通過微衛星識別工具MISA軟件對組裝基因組的Scaffolds進行SSR分析。4種貝類總共分別搜索到 928 562、824 342、739 299、739 407個SSR。其中,4種貝類分別有 196 158、127 288、158 667、160 694條序列包含1個以上堿基重復;分別有13 360、127 897、118 293、118 258 個復合形式存在。4種貝類SSR位點的核苷酸重復類型可劃分6種類型。其中,單核苷酸重復的SSR基元類型最豐富,五核苷酸重復、六核苷酸重復基元類型和分布相對較少。具體而言,中華園田螺、銅銹環棱螺、多棱角螺、湄公螺的單核苷酸重復、雙核核苷酸重復序列占比最高,分別達79.2%、79.9%、78.7%、80.9%(表4)。對檢測出的重復基元進行排序,其中,4種貝類優勢重復基元分別是A/T、AC/GT、AAT/ATT、AGAT/ATCT、ATAAT/ATTAT、CTAACC/GGTTAA。對雙核苷酸重復、三核苷酸重復、四核苷酸重復中基元進行比較發現,4種貝類的優勢重復基元大體相同(圖3)。
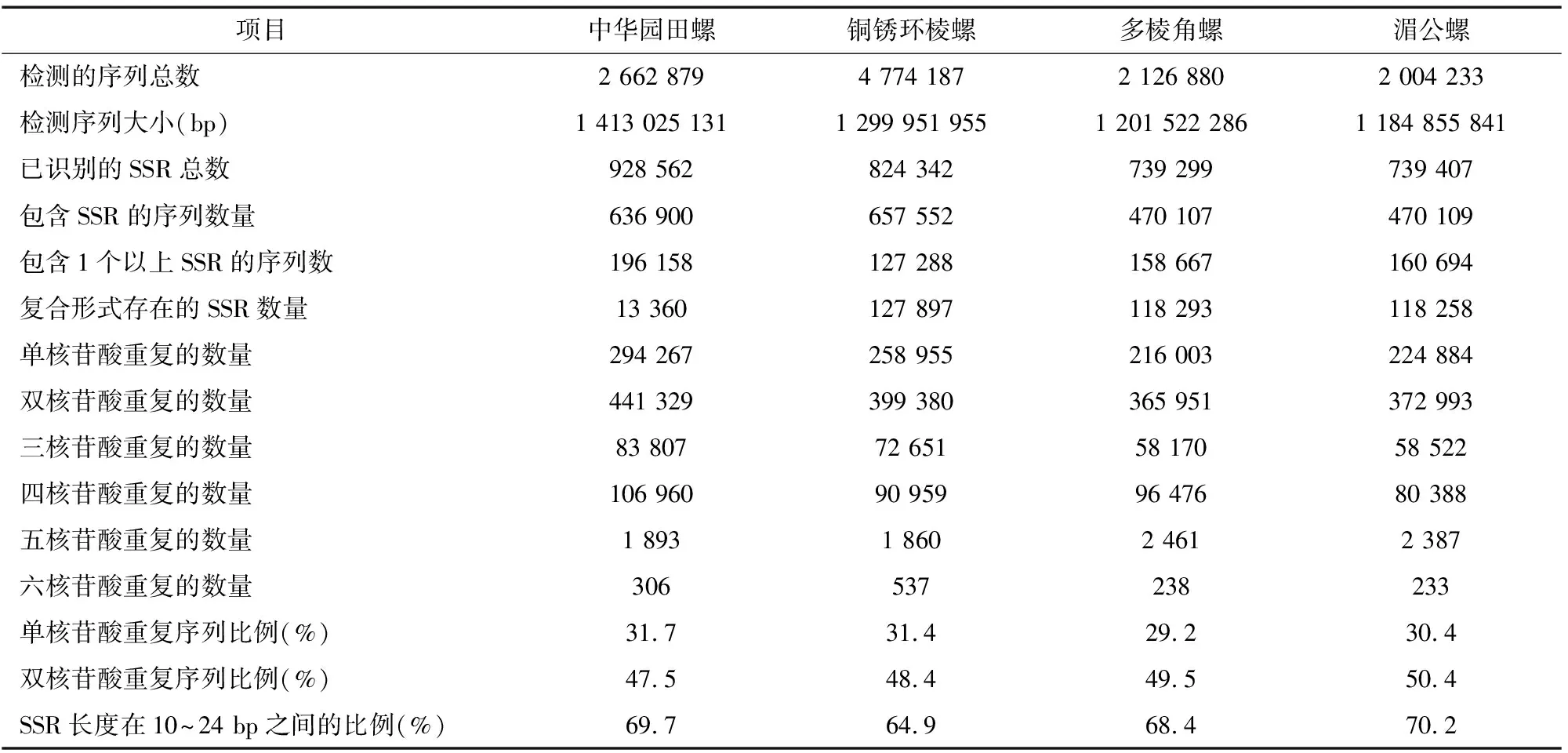
表4 田螺科4種螺基因組SSR類型

3 結論與討論
近年來,隨著測序技術的發展,越來越多的軟體動物基因組被解析,為研究軟體動物起源、進化、生殖、發育、性別調控和免疫等問題提供分子生物學基礎。目前基因組調研的主要手段是高通量二代測序技術。本研究通過Illumina數據,利用生物信息學手段,首次對腹足綱淡水軟體動物田螺科貝類中華園田螺、銅銹環棱螺、多棱角螺、湄公螺等代表性物種的基因組大小、雜合度進行評估分析。4種貝類的基因組大小分別為1 353、896、991、1 196 Mb,說明田螺科物種基因組大小存在差異,但與已公布大多數腹足綱物種的基因組大小接近[19-21]。腹足綱軟體動物基因組大小存在豐富的多樣性,如在已測腹足綱中,基因組最大的物種是地中海芋螺(Conusventricosus),基因組大小為 3.6 G;霸王蓮花青螺(Lottiagigantea)基因組最小(359.5 Mb)[8],這2個物種基因組大小差異約為10倍。基因組是反映生物物種遺傳信息的重要指標,在物種進化和適應中起著重要作用,其大小與生物參數有關,如細胞大小、抗逆性和個體生長等。在本研究中,田螺科貝類生長個體大小與基因組大小沒有直接的相關性。
前人將一個物種雜合度高于0.5%定義為高雜合度物種[22],在本研究中,中華園田螺、銅銹環棱螺、多棱角螺、湄公螺4種貝類的雜合度分別約0.77%、1.35%、2.41%、0.91%,均大于0.5%,說明這4種田螺科貝類是高雜合率物種,這可能與田螺科物種的特殊性有關。田螺科貝類屬于雌雄異體,且生境基本相同,不同種群間基因交流可能是造成雜合率高的原因之一。這在Wang等應用線粒體基因組構建系統發育樹的研究中有所顯示[23],說明田螺科存在著種間基因交流,而部分種間是否存在生殖隔離還存在爭議,需要進一步研究。實踐證明,高雜合度基因組會導致組裝過程中同源區域的read無法合并,分支結構過多,連續性降低,從而造成組裝的基因組偏大。在本研究中也存在這樣的問題,應用二代測序數據從頭組裝結果大小均高于應用K-mer評估的結果。另外,高雜合還導致組裝結果中Contig N50較小,造成GC平均深度及分布較小。以上結果說明高雜合度提高了對田螺科物種精細基因圖譜的構建難度。在本研究中4個田螺科物種重復序列占全基因組較小,分別為26.2%、26.7%、27.8%、28.8%,與已發表的其他腹足綱物種接近[24-26],為低重復序列基因組(重復序列比例高于50%是高重復基因組)[27],但非洲大蝸牛(Achatinafulica)例外,其重復序列比例較大,占整個基因組的70%,前人研究發現其發生基因組復制事件[28]。本研究4個田螺科貝類基因組重復序列占比較小,推測較少發生全基因組復制事件。
物種GC比例是評估、分析物種調研圖準確度,評估后續基因組圖譜組裝難度的重點衡量標準之一[29]。在本研究中,田螺科4種貝類的GC含量分別為33.4%、33.7%、33.1%、33.8%,腹足綱其他物種GC含量在26.8%~45.5%的區間內,其中綠唇鮑(Haliotislaevigata)GC含量最小(26.8%)[30],印度洋熱液噴口蝸牛(Alviniconchamarisindica)最大(GC含量為45.5%)[31]。事實上腹足綱物種線粒體基因組中也有存在GC含量較低的規律,如福壽螺(Pomaceacanaliculata)、斑點果瓶螺(P.maculat)和皺紋盤鮑(Haliotisdiscushannai)的線粒體GC含量在33%~40%范圍內[32],說明腹足綱物種核基因組和線粒體基因組均具有AT偏好性。研究發現物種在基因組圖譜構建中,過高或過低的GC含量將導致測序錯誤,GC貧乏或富集區通常會引起擴增效果較差,并影響組裝數據的準確性[33],且對準確度的干擾高于完整性。這也是本研究4個貝類物種中基因組初步組裝質量較差,以及其他貝類基因組序列組裝難度較高的因素之一[34]。
SSR分子標記具有多態性高、等位差異顯著的優點[35],是遺傳研究中應用廣泛的分子標記[36-37]。本研究利用MISA分析方法對田螺科代表性4種螺進行SSR特征分析,與植物的SSR主要是雙核苷酸和三核苷酸不同[22],本研究4種貝類中單、雙核核苷酸重復序列占比最高,分別達79.2%、79.9%、78.7%、80.9%,這與扁玉螺(Neveritadidyma)[35]、泥東風螺(Babylonialutosa)[38]的研究結果相一致。從4種田螺科貝類的SSR重復基元來看,單核苷酸中SSR位點占比最多的為A/T,雙核苷酸重復基元中,AC/GT占比均最高,這與前人研究的結果[39]相一致。SSR的長度會影響其本身多態性,而4種螺所含SSR的長度絕大部分在10~24 bp 之間的,分別占71.4%、64.9%、68.4%、70.2%,推測田螺科物種可能受到強烈的趨同選擇壓力。本研究為田螺科物種基因圖譜構建、物種分子鑒定提供了前期基礎。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2024年12期)2024-12-02 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
中學生博覽(2022年7期)2022-06-21 21:48:14
大科技·百科新說(2021年8期)2021-11-03 10:55:16
學苑創造·A版(2021年5期)2021-06-28 19:51:42
少兒美術(快樂歷史地理)(2020年9期)2020-03-19 05:10:56
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
