巨噬細胞與腫瘤治療的研究進展
2023-05-30 10:53:18張玉民綜述劉金劍劉鑒峰審校
河北醫科大學學報 2023年5期
關鍵詞:劑量
王 丹,張玉民(綜述),劉金劍,劉鑒峰(審校)
(中國醫學科學院放射醫學研究所放射醫學與分子核醫學重點實驗室,天津 300192)
腫瘤發生不僅取決于癌細胞的內在特性,還取決于癌細胞與腫瘤微環境(tumor microenvironment,TME)成分的相互作用。巨噬細胞是TME中最豐富的免疫細胞之一,通常具有2種極化狀態,可響應不同的刺激而分化為M1型和M2型[1]。在腫瘤發展的初始階段,巨噬細胞可以通過殺死腫瘤細胞直接促進抗腫瘤反應。由于腫瘤或Th2細胞開始主導TME,腫瘤相關巨噬細胞(tumor-associatd macrophage,TAM)開始表現出免疫抑制的M2型,通過產生大量生長因子、細胞外基質重塑分子和細胞因子調節腫瘤的生長、遷移和血管生成[2]。已有許多研究表明M2表型的巨噬細胞與肝癌、結腸癌、胰腺癌、甲狀腺癌和腦腫瘤等有關[3],這表明M1-M2型巨噬細胞極化可能在癌癥治療中發揮一定的作用。
1 巨噬細胞的極化分型
巨噬細胞具有很強的可塑性,并具有功能多樣性。最初認為巨噬細胞表現為M1表型參與抗腫瘤免疫,但在惡性腫瘤進展過程中顯示其可以促進癌癥的發生,刺激血管生成和抑制抗腫瘤免疫,這是由于巨噬細胞已經表現成M2表型所導致的[4]。巨噬細胞的表型可以通過TME中的一些因素例如調節性T細胞產生的免疫抑制細胞因子、趨化因子、腫瘤細胞產物來調控,也可以通過Th1和Th2輔助細胞的細胞因子庫改變。微生物刺激如脂多糖和Th1相關細胞因子如干擾素γ(interferon-γ,IFN-γ)可以將巨噬細胞極化為M1表型[5],并在促炎、殺死微生物和腫瘤抵抗過程中發揮一定的作用。M1型巨噬細胞有以下特征:包括抗原呈遞能力,高產量的白介素(interleukin,IL)-6、IL-12和IL-23,高產量的有毒中間體包括一氧化氮(nitric oxide,NO)、活性氧中間體和基質金屬蛋白酶12,如圖1所示[6]。研究顯示,在乳腺癌小鼠模型中,唑來膦酸修飾的納米顆粒有效抑制了破骨細胞形成,并同時誘導巨噬細胞向M1促炎表型極化[7]。Th2細胞因子如IL-4和IL-13可以使巨噬細胞極化為M2表型,M2型巨噬細胞在抗炎、組織修復重塑、寄生蟲清除、促進腫瘤生長和免疫調節過程中起作用。Haydar等[8]研究顯示阿奇霉素通過抑制STAT1和核轉錄因子(nuclear factor kappa B,NF-κB)信號通路使巨噬細胞極化為M2型,從而有效控制囊性纖維化患者的過度炎癥。根據誘導信號極化的方式,M2型巨噬細胞又可分為以下4種:M2a、M2b、M2c和M2d型。其中M2a型是由IL-4或IL-13誘導的,通過分泌細胞外基質促進組織修復;M2b型是通過暴露于免疫復合物和Toll樣受體(Toll-like receptors,TLRs)或IL-1受體的激動劑參與抗炎反應和免疫調節功能誘導的[9];M2c型由糖皮質激素和IL-10誘導抑制免疫反應和組織重塑。受TME影響的TAM在腫瘤的生長、侵襲和轉移中起著至關重要的作用。TAM是一種替代活化的M2型巨噬細胞。一些TAM看起來類似于M2b表型(IL-10高,IL-12低),而另一些在鼠類和人類腫瘤研究中已顯示其腫瘤壞死因子α(tumour necrosis factor-α,TNF-α)低,表型類似于M2c表型。但是,一些學者將TAM歸類為M2d巨噬細胞,其表達高水平的血管內皮生長因子和IL-10。由腺苷引起的M2d型巨噬細胞中,白血病抑制因子和IL-6被認為在誘導血管生成、調節腫瘤進展和增強腫瘤的存活方面發揮關鍵作用(圖2)。
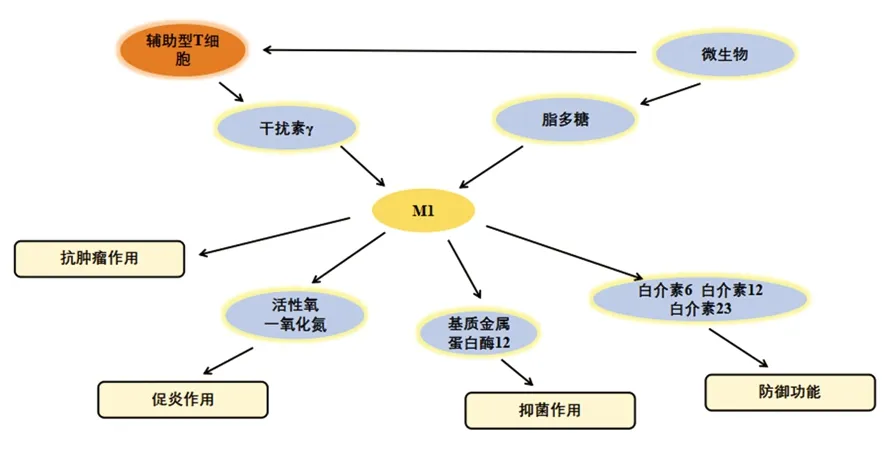
圖1 M1型巨噬細胞的激活因子和功能
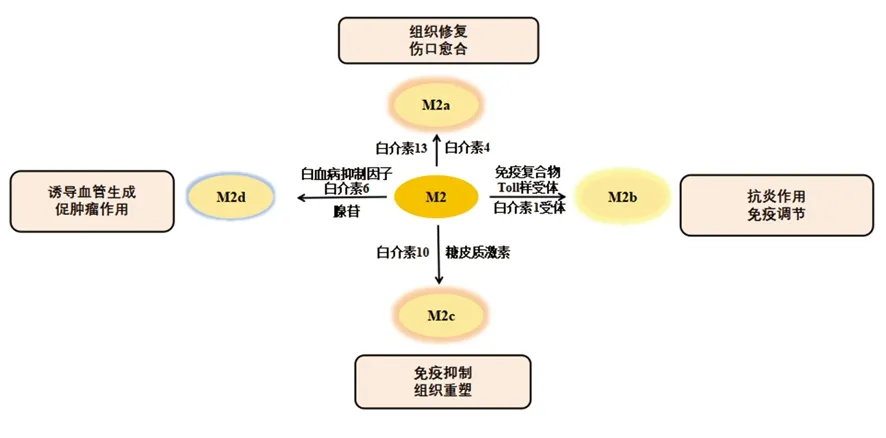
圖2 M2型巨噬細胞的激活因子和功能
2 巨噬細胞的表面標志物-細胞因子和趨化因子
循環單核細胞和組織巨噬細胞存在不同的受體和獨特的分泌模式見表1[6]。M1型巨噬細胞高度表達主要組織相容性復合體(major histocompatibility complex,MHC)Ⅱ、IL-1R、2、TLR4、CD80、CD86和其他刺激性分子。同時,M1型巨噬細胞分泌促炎細胞因子如TNF-α和IL-1,以及一些趨化因子如CCL2、CCL3、CCL5、CXCL8、CXCL9、CXCL10、CXCL11和CXCL16,會產生高水平的重要炎癥因子,包括IL-23、IL-6、IL-12[4]。M1型巨噬細胞也與活性氧(reactive oxygen species,ROS)的合成和NO的釋放有關。褐藻糖苷作為一種抗氧化劑,可抑制單核細胞/巨噬細胞和侵襲性癌細胞中的細胞內ROS和線粒體超氧化物水平。研究顯示,與ROS抑制劑相比,褐藻糖苷直接誘導單核細胞向M1型巨噬細胞極化,并將M2巨噬細胞重新極化為M1表型[10]。NO也在M1型巨噬細胞中扮演重要的角色。研究顯示,M1表型TAM負責誘導性一氧化氮合成酶的表達并催化NO的合成,釋放的NO進入TME通過作用于M1型巨噬細胞在腫瘤抑制中發揮作用[11]。此外,高通量的NO到腫瘤基質也刺激TAM向M1表型轉換[12],從而協調基質內的抗腫瘤信號。
M2型巨噬細胞也表達許多MHC-Ⅱ分子,但是這種表達不足以有效地呈遞抗原;其還表達高水平的精氨酸酶1,可以促進多胺的合成并刺激組織修復、細胞生長、膠原蛋白形成等。其可細分為M2a、M2b、M2c和M2d型。M2a型巨噬細胞除了產生CCL17、CCL18、CCL22和CCL24外,還表達高水平的表面分子和受體如CD163、CD23、CD209、Fizz1、Arg1、Ym1/2、IL-4R、FcR、CXCR1、CXCR2和Dectin-1等。M2b型巨噬細胞表達高水平的表面分子CD80和CD86,并產生TNF-α、CCL1、IL-1、IL-6和IL-10。M2c型巨噬細胞表達高水平的表面受體和分子,包括CD14、CD50、MR和SR,并產生IL-10、CCL16、CCL18、CXCL13和轉化生長因子β(transforming growth factor-β,TGF-β)。M2d型巨噬細胞(TAM)表達高水平的血管內皮生長因子和CD163,產生細胞因子如IL-10、IL-12、TNF-α和TGF-β,并分泌趨化因子CCL5、CXCL10、CXCL16、CCL18。

表1 巨噬細胞的表面標志物、細胞因子和趨化因子
3 M1-M2極化的分子途徑
巨噬細胞的極化受轉錄因子和miRNA驅動的幾種機制的影響如圖3。
3.1干擾素調節因子(interferon regulatory factor,IRF)/轉錄激活因子(signal transducer and activator of transcription,STAT)信號通路 巨噬細胞向M1或M2表型極化主要是由IRF/STAT信號通路誘導的。其中IRF3、IRF5、STAT1和STAT5負責驅動M1極化,而IRF4、STAT3和STAT6則提供M2表型的激活信號。IFN-γ是一種有效的內源性巨噬細胞激活因子,可以主要激活STAT1,并通過IFN-γ/JAK/STAT1通路發出信號誘導M1樣巨噬細胞極化[13]。除了STAT1信號通路,有研究顯示,通過阻止CrkL-STAT5復合物的形成,可以減輕M1表型轉換和巨噬細胞炎癥[14]。Th2細胞因子如IL-13和IL-4,其啟動子受信號轉導和STAT6調節,通過誘導過氧化物酶體增殖物激活受體表達在巨噬細胞中產生M2樣激活[15]。Wu等[16]研究顯示瑪格毒素通過抑制p-STAT1活性調節M1標志物的表達,并通過促進p-STAT6活性來增加M2標志物的表達。此外,IRF1、IRF5和IRF-8與M1表型極化相關,而IRF3和IRF4參與M2巨噬細胞的極化過程[17]。
3.2低氧誘導因子(hypoxia inducible factor,HIF)信號通路 除了IRF/STAT轉錄因子外,缺氧也會影響巨噬細胞的極化。缺氧能以不同的方式激活M1和M2巨噬細胞中的HIF。Th1細胞因子能夠誘導HIF-1α表達,引發M1應答;而Th2細胞因子在M2巨噬細胞中會活化HIF-2α。這些差異取決于HIF-1和HIF-2分別激活或抑制NO合成的能力。最近的一項研究表明,從低氧腫瘤區域釋放的外泌體而不是從常氧區域釋放的外泌體能促進M2型巨噬細胞極化[18]。具有抗腫瘤活性的M1型巨噬細胞停留在腫瘤中的顯示,表明可開發一種新穎的利用TAM對抗癌癥的治療方法。
3.3NF-κB信號通路 NF-κB在促炎性巨噬細胞扮演著重要角色。事實上,活化的異質二聚體NF-κB(p50-p65),促進了抗炎性巨噬細胞中炎癥基因的轉錄;而抑制異質二聚體NF-κB(p50-p50)則阻止了這些基因的轉錄。Liu等[19]研究顯示顆粒蛋白前體通過NF-кB和MAPK通路抑制LPS誘導的巨噬細胞M1極化。其他轉錄因子包括轉錄因子激活蛋白1、Kruppel樣因子4和過氧化物酶體增殖物激活受體也可以調節巨噬細胞的活化狀態。
3.4miRNA信號通路 miRNA也會干擾巨噬細胞的極化。miR127、miR155和miR223是M1極化的關鍵調控因子,而miR146a則促進了M2極化,降低了M1標記的表達。Wang等[20]研究顯示miR-99b的靶向遞送可重新編程與腫瘤相關的巨噬細胞表型,從而導致腫瘤消退。由于上述通路有時會相互作用,因此研究與M1和M2表型相關的巨噬細胞極化的信號通路在理解和創造預防腫瘤發展的治療方法方面起著重要的作用。
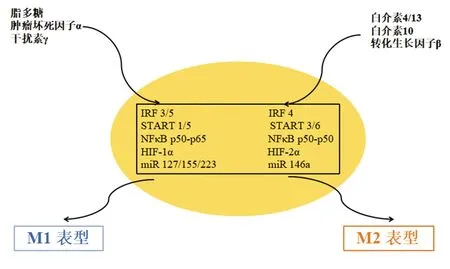
圖3 巨噬細胞向M1和M2極化的分子途徑
4 TAM與癌癥
TAM是腫瘤微環境中主要的浸潤性白細胞,并且在炎癥與癌癥之間發揮關鍵作用。在癌癥初期,TAM可能發揮強大的免疫活性;但在后期,TME通過富集生長因子和抗炎介質,使TAM向促進腫瘤生長的M2型極化。
TAM影響腫瘤進展的不同方面。首先,其可以調節細胞外基質降解的酶和蛋白酶如基質金屬蛋白酶、纖溶酶、骨連接蛋白和組織蛋白酶,TAM對細胞外基質的破壞促進了腫瘤細胞的擴散和轉移。其次,TAM也導致腫瘤內的免疫抑制環境,TAM通過抑制抗原呈遞細胞,使其無法分泌IL-12,并且產生高水平的IL-10和TGF-β,從而阻斷T細胞增殖,抑制細胞毒性T淋巴細胞(cytotoxic T lymphocyte,CTL)反應并激活Treg[21]。作為對微生物信號、組織損傷、細胞因子和代謝產物的響應,組織中的單核細胞和巨噬細胞能夠進行重新編程,實現M1與M2表型之間的轉化。TAM促進腫瘤生長的性質及其從M2型巨噬細胞向M1表型轉變的能力,使其成為一個有吸引力的抗癌治療靶點。通過靶向TAM,許多研究在不同的腫瘤模型中均取得了良好的抗腫瘤作用。目前,放射療法、化學療法和免疫療法仍然是臨床上腫瘤治療的重要方式。然而,由于治療耐藥性或放療和化療過程中的不良反應,一些接受治療的患者仍然患有腫瘤轉移或局部復發性疾病。因此,靶向TAM的藥物與其他癌癥治療方法相結合的策略將提供一種有希望的手段。
4.1靶向TAM的化學療法 雖然化療藥物的開發主要是為了誘導快速分裂的癌細胞死亡,但許多藥物對非癌細胞群體也有藥理作用。有研究表明氯膦酸鹽脂質體耗盡巨噬細胞可阻止骨髓瘤的發展[22],其發生機制為含有氯膦酸鹽的脂質體被巨噬細胞吞噬后,經過溶酶體處理破壞氯膦酸鹽在細胞中釋放,隨后氯膦酸鹽代謝成類似于ATP的物質對巨噬細胞產生細胞毒性進而抑制骨髓瘤的發展。
除了發揮細胞毒作用外,幾種化療藥物還能調節巨噬細胞極化的表型。2個主要的轉錄因子(STAT3和STAT6)已大量報道參與阻斷M2表型。白藜蘆醇通過抑制STAT3通路實現對TAM向M2型極化的抑制,進而成功的抑制了小鼠肺癌異種移植模型中的腫瘤生長,其阻斷TAM的M2樣極化與STAT3活性降低有關[23]。類似地,芬維A胺抑制STAT6的磷酸化實現對TAM向M2極化的有效抑制,在結腸直腸小鼠模型中,這種效應伴隨著腫瘤中M2樣巨噬細胞數量的減少和血管生成的抑制[24]。此外,其他STAT6激活的抑制劑(TMC264,AS1517499)也被開發出來,但尚未進行臨床研究。紫杉醇作為一種常用的抗腫瘤化療藥物,有研究顯示其通過將與腫瘤相關的巨噬細胞重編程為TLR4依賴性的M1型,從而抑制了腫瘤的生長[25]。為了對抗這些耐藥機制,化療和免疫治療的結合可能會使腫瘤消退更有效。值得注意的是,紫杉醇和吉西他濱是2種化療藥物,其可能誘導免疫原性死亡,通常與其他TAM調節治療藥物聯合治療。
4.2靶向TAM的放射療法 放射療法是一種主要用于惡性腫瘤的局部治療形式。許多研究顯示放射療法會改變巨噬細胞的表型,這為癌癥的治療提供了一個新的思路。Stary等[26]研究顯示直腸癌患者的短程放療與TAM向M1樣促炎表型的轉變有關。但是,關于放療對巨噬細胞表型的影響有矛盾的結果。有研究表明,腫瘤照射與TAM的免疫抑制表型有關[27]。Seifert等[28]研究顯示與未放射的小鼠相比,來自放射治療的浸潤性和浸潤前胰腺腫瘤的巨噬細胞比例更大,具有免疫抑制性的M2樣表型。這提示放療的某些方面會影響巨噬細胞的表型。因此,為了分析放療對巨噬細胞重編程的影響,Genard等[29]根據放射劑量進行了分類(低劑量為<1 Gy的劑量,中劑量為1~10 Gy的劑量,高劑量為>10 Gy的劑量),顯示高劑量和低劑量對巨噬細胞從M2向M1的極化無影響,而中劑量明顯引起TAM重編程。同時在進行全身放療的實驗中顯示:健康小鼠在接受低劑量或中劑量的全身輻照后,巨噬細胞均趨向M1表型。顯然,輻照的效果似乎取決于腫瘤模型,輻照劑量和組織等多種因素。
至于不同放射劑量導致TAMs的表型不同,是由于不同劑量的放療后,p50-p50和p65-p502個二聚體組成的NF-κB平衡狀態的不同[29]。照射劑量對巨噬細胞NF-κB平衡的影響相反:低劑量或1Gy以下劑量均不改變照射后巨噬細胞核p50-p50 NF-κB的豐度。中劑量或1-10 Gy劑量誘導NF-κB平衡從非活性同源二聚體(p50-p50)切換到活性異源二聚體(p50-p65),一旦p50-p65被轉錄到核內,促炎因子(TNF-α、IL-10、IL-6)等就會高表達,進而使巨噬細胞表現為M1表型。高劑量或高于10 Gy的劑量不能改變NF-κB的平衡,使得TAM傾向于M2表型。
基于放療對TAM的極化影響,山東師范大學唐波教授課題組開發了X射線誘導表型轉化策略[30],將工程化修飾的、M0代的巨噬細胞在體外經X射線照射之后,改造成具有激活腫瘤免疫的M1型TAMs。體內外試驗證實了X射線誘導表型轉化策略能夠將巨噬細胞改造成M1型,進而顯著抑制小鼠腫瘤的生長。因此,巨噬細胞極化策略將有助于增強臨床上放射治療或放療聯合免疫治療的抗腫瘤效率。
4.3靶向TAM的免疫療法 在過去的幾年中,已經開發了多種腫瘤學免疫策略來重新激活適應性免疫和先天免疫系統,以建立強大的抗腫瘤免疫應答。作為經典抗癌治療的替代方法,腫瘤通常會對此產生抗藥性。免疫檢查點抑制劑——針對細胞毒性T淋巴細胞抗原4的單克隆抗體,可增強細胞毒性CD8+T細胞活性的程序性細胞死亡受體1和程序性死亡受體-配體1的臨床試驗已顯示成功地治療了黑色素瘤和肺癌。但是,在大多數情況下,出于未知原因只有一小部分患者對免疫療法完全有效。
目前已經顯示了幾種常用于將TAM從M2型重新編程為M1表型的藥物,主要包括TLR激動劑、靶向M1表型的抑制蛋白單克隆抗體以及其他化合物。TLR激動劑是一種很有前途的抗腫瘤藥物,已有研究顯示包裹CpG寡脫氧核苷酸的靶向鐵蛋白納米顆粒可以誘導TAM從M2表型極化為M1表型,并抑制腫瘤生長[31]。此外,研究顯示R848能夠通過巨噬細胞極化下調腫瘤免疫抑制微環境中髓源抑制性細胞的表達,進而增強了聯合治療中奧沙利鉑的抗腫瘤作用[32]。
另一種有利于TAM細胞毒功能的方法是用單克隆抗體刺激CD40。抗CD40單抗可以促進巨噬細胞的抗腫瘤作用,特別是通過加強NO和TNF-α的分泌,因此可以誘導CD8+T細胞抑制腫瘤的生長和轉移。抗CD40單抗可在TAM和腫瘤浸潤性單核細胞質膜中誘導PD-L1上調,同時有研究顯示PD-L1軸的阻斷與抗CD40和抗CTLA-4 單抗在結腸癌和乳腺癌模型中顯示出廣泛的存活率[33]。
重編程TAM的第三個替代方法是使用不同的化合物。最著名的一種是FDA批準的IFN-γ。研究顯示白藜蘆醇類似物的治療顯著增加了脾細胞中IFN-γ分泌細胞的表達并減少了M2型巨噬細胞的浸潤,誘導的IFN-γ逆轉了TAM的特性,促使TAM重編程為M1型[34]。不同的納米粒子(nanoparticles,NPs)如金屬NPs、氧化物NPs、質粒DNA NPs等,可以誘導M0巨噬細胞向各種表型極化并調節巨噬細胞重編程。Chattopadhyay等[35]研究顯示抗原鍵合修飾的氧化鈷NPs通過調節NADPH氧化酶和p38MAPK將TAM重編程為促炎表型。除此之外,還顯示了一種小的類黃酮化合物DMXAA,既是血管破壞劑,也是干擾素基因刺激因子的鼠激動劑。可以使M2表型的巨噬細胞重新極化,從而發揮腫瘤的免疫治療[36]。其發揮作用的機制是利用DMXAA激活干擾素基因刺激因子成為二聚化的結構,隨后進一步與環鳥腺苷酸小分子結合,構象發生改變,招募TANK結合激酶1蛋白,促使其磷酸化IRF3,磷酸化的IRF3入核,誘導I型干擾素的產生,進而調控巨噬細胞向M1型極化。因此,利用TAM重編程的策略調控免疫治療會在一定程度上更有效地抑制腫瘤生長和轉移。
5 結論與展望
越來越多的研究已經顯示巨噬細胞極化在癌癥的發生發展中發揮重要作用。由于化學療法、免疫療法和放射療法的局限性,利用TAM重編程的能力將靶向TAM與這三種療法結合將為進一步提升癌癥治療的精準性提供可能。TAM重編程或者靶向TAM治療是通過哪些機制實現對化學療法、免疫療法和放射療法抗腫瘤效率的提升,需要具體的研究以期為癌癥的臨床治療提供更有效的治療思路和方法。
猜你喜歡
課堂內外·初中版(科學少年)(2023年10期)2023-12-10 00:43:06
全科護理(2022年10期)2022-12-26 21:19:15
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
今日農業(2022年4期)2022-11-16 19:42:02
鄉村科技(2021年33期)2021-03-16 02:26:54
國際放射醫學核醫學雜志(2021年10期)2021-02-28 08:41:58
藥學與臨床研究(2015年4期)2015-06-05 11:35:54
衛生職業教育(2014年24期)2014-05-20 09:05:38
同位素(2014年2期)2014-04-16 04:57:20
中國合理用藥探索(2014年11期)2014-03-11 20:30:20
