白果仁及炒白果仁內源性有害成分分析
2023-06-04 07:00:14張雁淼丁明和談仲川周天天干國平1b
湖北農業科學 2023年5期
張雁淼,丁明和,張 營,談仲川,周天天,干國平,1b
(1.湖北中醫藥大學,a.藥學院;b.湖北省中藥炮制工程技術研究中心,武漢 430065;2.馬應龍藥業股份有限公司,武漢 430064)
白果為銀杏科植物銀杏(Ginkgo bilobaL.)的干燥成熟種子,藥食同源[1]。中國白果資源豐富,年產量約2 萬t[2]。白果中含有銀杏葉黃酮內酯[3]、多元醇等活性成分,并含有豐富的多糖[3,4]、蛋白質、淀粉、脂肪、維生素C、氨基酸和礦物質等營養成分[4],具有良好的臨床療效和食用價值[5-7]。但長期以來,由于白果有一定毒性,嚴重制約了白果資源的開發利用[8]。現代研究表明,白果中內源性有害成分為總銀杏酸、吡哆醇、氰苷。總銀杏酸為水楊酸的6-烷基或6-烯基衍生物,主要由銀杏新酸、銀杏酸、氫化銀杏酸、氫化銀杏酸、銀杏二酚等組成,具有細胞毒性、神經毒性、免疫毒性[9],含量過高會導致腹痛、腹瀉、過敏性皮炎等不良反應[10]。吡哆醇主要由4′-O-甲基吡哆醇(MPN)、4′-O-甲氧基吡哆醇-5′-葡萄糖苷(MPNG)等組成[11],統稱為銀杏毒(TMPN),含量過高會引起嘔吐、腹瀉、抽搐癲癇等[12]。氰苷成分通過體內代謝分解為氫氰酸,氫氰酸可導致細胞缺氧死亡引起呼吸加快加深、心律不齊、脈搏加快、抽搐昏迷,最后意識喪失,呼吸衰竭而亡[13]。
白果生食有毒,中醫認為炒制可降低毒性[14],但未見炒制白果可降低毒性的關鍵證據。雖為藥食同源品種,卻有食用白果和食用炒白果中毒的報道[15]。迄今為止,未見對白果及其飲片內源性有害成分的分析研究,《中國藥典》中亦未對白果仁及其炮制品炒白果仁中內源性有害成分進行含量控制和安全性評價,因此無法保證白果及其飲片的安全性。本研究采用HPLC 法、HS-GC 法分別建立白果仁中總銀杏酸、吡哆醇、氰苷的含量分析方法,為白果及其飲片的質量控制、安全用藥和白果的開發利用提供科學依據。
1 材料與方法
1.1 材料與試劑
10 批白果藥材分別采自湖北省安陸市、湖北省巴東縣、湖北省武漢市黃陂區、江蘇省徐州市邳州市、江蘇省泰興市新街鎮、河南省信陽市新縣、山東省臨沂市郯城縣、重慶市涪陵區、云南省保山市騰沖市、廣西壯族自治區桂林市靈川縣(樣品編號依次為1 號至10 號)。經湖北中醫藥大學湖北省中藥炮制工程技術研究中心鑒定為銀杏科植物銀杏的種子。標本保存于湖北中醫藥大學藥物化學研究室1 室。
供試品白果仁(生品)處理:白果去殼,參照《中國藥典》[16]撣法(通則0213)去內種皮;供試品炒白果仁(炒制品)處理:參照《中國藥典》[16]清炒法(通則0213)制備,白果仁、炒白果仁均經粉碎(過3 號篩)處理。
白果新酸對照品、總銀杏酸對照品,成都埃法生物科技有限公司;MPN 對照品,成都普思生物科技股份有限公司;MPNG 對照品,自制,HPLC 歸一化法測定純度為98.7%;水中氰成分分析標準物質,GBW(E)080115,中國計量科學研究;甲醇,色譜純,常熟市鴻盛精細化工有限公司;乙腈,色譜純,西格瑪奧德里奇(上海)貿易公司;氯胺T,分析純,國藥集團化學試劑有限公司;磷酸、鹽酸、氫氧化鈉、甲基橙、三氟乙酸均為分析純。
1.2 儀器與設備
LC-20AD 型高效液相色譜儀(配有SPD-20A 型紫外可見光檢測器及LabSolutions 數據處理軟件),日本島津公司;Aglient7890B 型氣相色譜儀(配有電子捕獲檢測器)、Aglient7697A 型頂空進樣器,美國安捷倫科技有限公司;AS 系列超聲波清洗機,天津奧特賽恩斯儀器有限公司;BS210S 型十萬分之一天平,北京賽多利斯天平有限公司;hhS-2S 型電子恒溫水浴鍋,上海康華生化儀器制造有限公司。
1.3 色譜條件
1)總銀杏酸色譜條件。
色譜柱為YMC-Triart C18(4.6 mm×150 mm,5 μm),以0.1%三氟乙酸的乙腈溶液為流動相A,以0.1%三氟乙酸的水溶液為流動相B,進行梯度洗脫(表1)。柱溫為30 ℃,檢測波長為310 nm,流速為1.0 mL/min,進樣量為20 μL。

表1 總銀杏酸流動相洗脫程序
2)MPN 和MPNG 色譜條件。
色譜柱為YMC -Triart C18(4.6 mm×150 mm,5 μm);以甲醇為流動相A,以去離子水為流動相B,進行梯度洗脫(表2)。柱溫為30 ℃,檢測波長為328 nm,流速為1.0 mL/min,進樣量為10 μL。

表2 MPN 和MPNG 流動相洗脫程序
3)氫氰酸色譜條件。
色譜柱為DB-WAX 毛細管柱(30 m×0.25 mm,0.25 μm);色譜柱溫度:40 ℃保持5 min,以50 ℃/min速率升至200 ℃,保持2 min;載氣:N2;進樣口溫度:200 ℃;檢測器溫度:250 ℃;尾吹:60 mL/min;分流比:5∶1;柱流速:2.0 mL/min。頂空平衡溫度:50 ℃;平衡時間:30 min。
1.4 溶液的制備
1)總銀杏酸。
對照品溶液:精密稱取白果新酸對照品2.03 mg,置于50 mL 容量瓶中,加甲醇溶解并稀釋至刻度,搖勻(濃度為40.60 μg/mL)。
定位用對照品溶液:精密稱取總銀杏酸對照品1.08 mg,置于10 mL 容量瓶中,加甲醇溶解并稀釋至刻度,搖勻,即得(濃度為108.00 μg/mL)。
供試品溶液:取樣品粉末4.00 g,精密稱定,置于具塞錐形瓶中,精密加入甲醇20 mL,稱定重量,水浴回流3 h,放冷至室溫,用甲醇補足減失的重量,搖勻,用微孔濾膜過濾,取續濾液,即得。
2)MPN 和MPNG。
對照品溶液:精密稱取MPN 對照品4.95 mg,置于50 mL 容量瓶中,加入甲醇溶解并稀釋至刻度,搖勻(濃度為99.00 μg/mL)。精密稱取MPNG 對照品5.60 mg,置于50 mL 容量瓶中,加入甲醇溶解并稀釋至刻度,搖勻(濃度為112.00 μg/mL)。
供試品溶液:取樣品粉末1.00 g,精密稱定,置于具塞錐形瓶中,精密加入去離子水50 mL,稱定重量,超聲30 min,放冷,用去離子水補足減失的重量,離心10 min,用微孔濾膜過濾,取續濾液,即得。
3)氫氰酸。
對照品溶液:精密吸取水中氰成分分析標準物質2 mL,置10 mL 容量瓶中,用0.1%氫氧化鈉溶液定容(濃度為10 mg/L),用去離子水稀釋,配制濃度為0、0.001、0.002、0.010、0.050、0.100 mg/L 的氰離子(以CN-計)標準工作溶液。
供試品溶液:取樣品粉末約1.00 g,精密稱定,置100 mL 容量瓶中,加去離子水適量,超聲30 min,放冷,定容至刻度,4 000 r/min 離心5 min,精密吸取上清液10 mL,置頂空瓶中,加16.7% 磷酸溶液0.2 mL,渦旋混合,加氯胺T 溶液0.2 mL,立即加蓋密封,渦旋混合,即得。
1.5 定量限與檢測限
精密吸取對照品溶液,用甲醇逐級稀釋至信噪比S/N=10 時即為定量限,至信噪比S/N=3 時即為檢測限。
1.6 標準曲線繪制
1)總銀杏酸標準曲線。
分別精密吸取白果新酸對照品溶液(濃度為40.60 μg/mL)1、2、3、4、5 mL,加入甲醇定容至10 mL容量瓶,混勻,制成一系列不同濃度的溶液,進樣分析。以峰面積(y)對溶液濃度(x)進行線性回歸。
2)MPN 和MPNG 標準曲線。
分別精密吸取MPN 對照品溶液(濃度為99.00 μg/mL)120、160、200、240、280 μL,加入甲醇定容至10 mL,混勻,制成一系列不同濃度的溶液,進樣分析。精密吸取MPNG 對照品溶液(濃度為112.00 μg/mL)0.6、0.8、1、1.2、1.4 mL,加入甲醇定容至10 mL,混勻,制成一系列不同濃度的溶液,進樣分析。以峰面積(y)對溶液濃度(x)進行線性回歸。
3)氫氰酸標準曲線。
分別取0、0.001、0.002、0.010、0.050、0.100 mg/L氰離子標準工作溶液各10.0 mL,置于6 個頂空瓶中,加入0.2 mL 磷酸溶液,渦旋混合,然后加入0.2 mL 氯胺T 溶液,立即加蓋密封,渦旋混合,進樣分析。以峰面積(y)對溶液濃度(x)進行線性回歸。
1.7 精密度試驗
按“1.4”項方法制備6 份供試品溶液,進樣分析,測定其峰面積,計算精密度。
1.8 溶液的穩定性試驗
分別取同濃度的供試品溶液和同濃度的對照品溶液,置于室溫下,于0、2、4、6、8 h 分別進樣分析。
1.9 回收率試驗
1)總銀杏酸。
精密稱取樣品6 份,每份2.00 g,置于具塞錐形瓶中,精密稱取白果新酸對照品1.827 mg,置于200 mL 容量瓶中,加甲醇溶解并稀釋至刻度,即得白果新酸對照品溶液(9.135 μg/mL),加入上述溶液20 mL,添加量分別為含藥量的50%,按“1.4”項下供試品溶液處理方法制備,進樣分析,計算回收率。
2)MPN 和MPNG。
精密稱取樣品6 份,每份0.50 g,置于具塞錐形瓶中,分別精確吸取MPN 對照品溶液(濃度為99.00 μg/mL)、MPNG對照品溶液(濃度為112.00 μg/mL)各4、8 mL,置于200 mL 容量瓶中,加甲醇溶解并稀釋至刻度,搖勻,即得MPN 對照品溶液(濃度1.98 μg/mL)、MPNG 對照品溶液(濃度4.48 μg/mL),加入上述溶液各25 mL,添加量分別為含藥量的50%,按“1.4”項下供試品溶液處理方法制備,進樣分析,計算回收率。
2 結果與分析
2.1 總銀杏酸的測定
2.1.1 系統適用性與專屬性 精密吸取總銀杏酸的對照品溶液、定位用對照品溶液、供試品溶液各20 μL,進樣分析。結果(圖1)表明,總銀杏酸供試品溶液色譜中可檢出除白果新酸外的4 種銀杏酸成分,且4 種銀杏酸成分峰與其他成分峰之間的分離度均大于1.5。

圖1 白果中總銀杏酸色譜
2.1.2 定量限與檢測限 按照“1.5”項方法測定,總銀杏酸的定量限為12.18 ng,檢測限為4.87 ng。
2.1.3 標準曲線繪制 按照“1.6”項方法測定,總銀杏酸回歸方程為y=10 218x+836.28,r=0.999 3,表明在濃度為0.609~20.30 μg/mL 時線性關系良好。
2.1.4 精密度試驗 按照“1.7”項方法測定,6 份供試品溶液總銀杏酸平均含量為90.08 μg/g,RSD=5.12%,表明該方法精密度良好。
2.1.5 溶液的穩定性試驗 按照“1.8”項方法測定,供試品溶液總銀杏酸峰面積的RSD為2.66%、對照品溶液白果新酸峰面積的RSD為3.79%,表明供試品溶液和對照品溶液在8 h 內穩定。
2.1.6 回收率試驗 按照“1.9”項方法測定,總銀杏酸的平均回收率為101.17%,RSD為4.57%(n=6)(表3),表明該方法測定結果準確、可靠。

表3 總銀杏酸的回收率試驗結果
2.2 MPN 和MPNG 的測定
2.2.1 系統適用性與專屬性 精密稱取MPN 對照品溶液、MPNG 對照品溶液、定位用對照品溶液、供試品溶液各10 μL,進樣分析。結果(圖2)表明,供試品溶液色譜中,MPN 峰、MPNG 峰與其他成分峰之間的分離度均大于1.5。

圖2 白果中MPN、MPNG 色譜
2.2.2 定量限與檢測限 按照“1.5”項方法測定,MPN 的定量限為5.94 ng,檢測限為2.38 ng,MPNG的定量限為11.20 ng,檢測限為4.48 ng。
2.2.3 標準曲線繪制 按照“1.6”項方法測定,MPN的回歸方程為y=17 070x+22 618,r=0.999 8,表明在濃度為1.188~2.772 μg/mL 時線性關系良好;MPNG的回歸方程為y=5 444.7x-202.79,r=0.999 8,表明在濃度為1.12~15.68 μg/mL 時線性關系良好。
2.2.4 精密度試驗 按照“1.7”項方法測定,6 份供試品溶液中MPN 的平均含量為110.36 μg/g,RSD=2.21%;MPNG 的平均含量為242.19 μg/g,RSD=2.57%,表明該方法精密度良好。
2.2.5 溶液的穩定性試驗 按照“1.8”項方法測定,供試品溶液MPN、MPNG 峰面積的RSD分別為0.50%、1.87%,對照品溶液MPN、MPNG 峰面積的RSD分別為0.40%、1.61%,表明供試品溶液和對照品溶液在8 h 內穩定。
2.2.6 回收率試驗 按照“1.9”項方法測定,MPN、MPNG 的平均回收率為103.14%、103.54%,RSD分別為4.80%、3.05%(n=6)(表4),表明該方法測定結果準確、可靠。
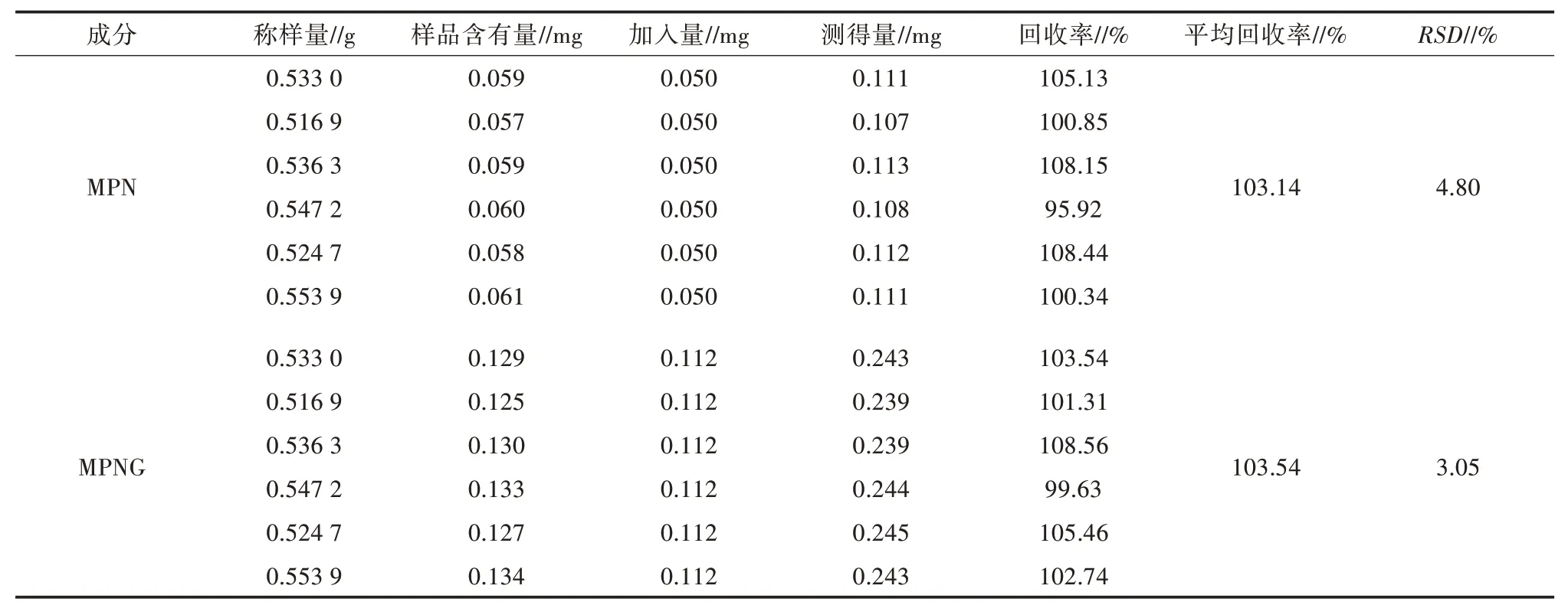
表4 MPN、MPNG 的回收率試驗結果
2.3 氫氰酸的測定
2.3.1 系統適用性與專屬性 頂空進樣分析氫氰酸的空白溶液(濃度為0 mg/L)、對照品溶液(濃度為0.001 mg/L)、供試品溶液。結果(圖3)表明,氯化氰與其他成分峰之間的分離度大于1.5,空白溶液無干擾。

圖3 白果中氰苷含量測定色譜
2.3.2 樣品提取時間和衍生化酸度的選擇 以去離子水為溶劑,樣品分別采用不同時間(20、30、40 min)進行超聲波提取。結果表明,超聲波處理30 min 時被測成分可提取完全。
氰化物衍生化需在弱酸性條件下進行,因此分別向頂空瓶中加入100、200、300、400 μL 磷酸溶液來考察酸度條件對衍生化反應的影響。結果表明,加入16.7%磷酸溶液300 μL 時,衍生化反應完全,被測成分峰面積達到最大值。
2.3.3 平衡溫度和平衡時間的選擇 分別采用不同平衡溫度(45、50、55 ℃),不同平衡時間(20、30、40 min),結果表明,當平衡溫度為50 ℃、平衡時間為30 min 時被測成分峰面積達到最大值。
2.3.4 定量限與檢出限 按照“1.5”項方法測定,氫氰酸定量限為10 ng,檢測限為3.5 ng。
2.3.5 標準曲線繪制 按照“1.6”項方法測定,氫氰酸的線性方程為y=9 770.7x+6.062 3,r=0.999 8,n=6,表明濃度為0~0.1 μg/mL 時線性關系良好。
2.4 不同產地的白果仁、炒白果仁中內源性有害成分分析
總銀杏酸成分存在于銀杏葉及其果實中。《中國藥典》對銀杏葉中總銀杏酸作了限度要求(總銀杏酸不得超過10 μg/g),但對白果及其炮制品中總銀杏酸未進行限度規定。由表5 可知,10 批不同產地的白果仁中總銀杏酸含量為48.597~229.822 μg/g,炒白果仁中總銀杏酸含量為44.973~151.058 μg/g。雖然經炮制處理后的各樣品中總銀杏酸含量與白果仁比較,均有所降低,但殘留量還是超過了銀杏葉中總銀杏酸的限度要求。

表5 不同產地的白果仁、炒白果仁中內源性有害成分的含量(單位:μg/g)
10 批不同產地白果仁中MPN 的含量為19.363~124.782 μg/g,MPNG的含量為265.896~814.585 μg/g,MPNG 含量遠高于MPN。炒白果仁中MPN 的含量為18.773~125.505 μg/g,與白果仁比較,7 個樣品的MPN 含量均減少,3 個樣品(2 號、3 號、5 號)的MPN含量升高;MPNG 的含量為24.646~687.892 μg/g,MPNG 含量均有所減少。但炒白果仁中MPN 和MPNG 的總含量仍有102.729~688.850 μg/g,表明白果通過傳統的炮制方法難以達到明顯的減毒效果。
歐盟第88/388/EEC 號指令規定灌裝果核飲料中氫氰酸限度為5 mg/kg。10 批不同產地白果仁中氫氰酸含量為0~0.176 μg/g,遠低于歐盟標準,說明白果中氰苷成分不是主要內源性有害成分。
2.5 不同產地、不同炮制方法白果仁中內源性有害成分的主成分分析
本研究使用Origin 軟件對10 批不同產地白果仁、炒白果仁中內源性有害成分的含量進行主成分分析(Principal Component Analysis,PCA)(圖4)。特征值大于1 的主成分(PC)分別為PC1(46.3%)、PC2(25.7%)和PC3(21.2%),由于白果仁中MPNG 的含量最高,所以白果仁的位置位于圖4a 的右上方,炒白果仁的位置位于下方,可能是由于處理后總銀杏酸和MPNG 的含量均有所降低。炒制處理后較高的MPN 保留量決定了樣品1 號、2 號和3 號的位置。從PCA 分析看出,白果仁和炒白果仁具有差異性。圖4b 中樣品4 號、5 號、6 號、7 號具有相近的MPNG 含量,樣品8 號、9 號具有相近的總銀杏酸含量。由于樣品9 號總銀杏酸含量最高,所以位于圖4b 的最上方,樣品1 號MPN 和氰苷含量最高,所以位于圖4b的左上方。總之,PCA 分析結果表明不同產地之間白果仁內源性有害成分的含量具有一定的相似性和差異性。

圖4 PCA 分析的載荷和得分
3 小結與討論
氰苷成分在人體中可代謝為氫氰酸,花彤彤等[17]認為白果中氰苷成分為主要內源性有害成分。氰化物的測定方法有分光光度法、氣相色譜法和苦味酸試紙法。苦味酸試紙法靈敏度低,分光光度法本身沒有分離功能且常受其他成分干擾,氣相色譜法分離能力強、檢測靈敏度高;倪洋[18]采用酸水解異煙酸-巴比妥酸法在全白果和白果胚乳中檢測到微量的氰化物;楊劍婷等[19]采用普魯士藍法、苦味酸試紙法和硝酸銀滴定法對白果胚和胚乳進行氰化物的測定,均未檢測到氰化物;本研究采用靈敏度高的頂空氣相色譜法測定。MPN 和MPNG 均具有神經毒性,是白果的主要有害成分。Kobayashi 等[20]、Chang 等[21]認為白果中主要的有害成分為MPN,炒白果仁中主要的有害成分為MPNG,MPNG 毒性小于MPN 的毒性,通過炮制可以降低白果毒性;單舒筠[22]報道白果炮制后MPN 含量降低,與本研究結果一致。
白果雖為藥食同源品種,但不同產地白果仁中內源性有害成分(吡哆醇和總銀杏酸)含量高、差別大,傳統炮制方法制得的炒白果仁中仍含有大量的內源性有害成分,臨床應用和食用均存在嚴重的安全隱患,建議國家主管部門重新審視白果的藥食同源屬性,同時根據白果內源性有害成分的性質及其毒性作用機理,加強采用現代炮制技術對白果進行炮制減毒的研究,在《中國藥典》白果及其飲片質量標準中增加內源性有害成分的檢查項目并作限度規定。
本研究建立了有效檢測白果仁、炒白果仁中內源性有害成分的方法,并對方法的可靠性性和檢出能力進行評價。結果表明,該方法專屬性強、重復性好,可有效進行質量控制,為白果的食用和臨床使用提供依據。
