以先天性甲狀腺功能減退癥及矮小為首發(fā)癥狀的假性甲狀旁腺功能減退癥1例報道并文獻復習
2023-06-28 07:10:04孟令哲初國銘張丹辛穎
中國醫(yī)科大學學報 2023年6期
關鍵詞:功能
孟令哲,初國銘,張丹,辛穎
(中國醫(yī)科大學附屬盛京醫(yī)院 1.小兒內(nèi)分泌科;2.遺傳科,沈陽 110004)
假性甲狀旁腺功能減退癥(pseudohypoparathyroidism,PHP)是由于分子缺陷致G蛋白(Gsα)α-亞基與受體結合異常,腺苷酸環(huán)化酶活化減弱,細胞內(nèi)信號傳導異常,從而引起靶細胞對促甲狀旁腺激素抵抗,部分還可合并對其他激素(如甲狀腺激素、促生長激素釋放激素、促性腺激素等)的抵抗,造成Albright遺傳性骨營養(yǎng)不良癥(Albright hereditary osteodystrophy,AHO)畸形、智力異常等[1]。本研究報道了1例先天性甲狀腺功能減退癥及矮小為首發(fā)癥狀的PHP1a型患兒,分析其臨床及遺傳學資料,并復習相關文獻,以期為PHP的臨床診療提供參考。
1 材料與方法
1.1 臨床資料
患兒,女,6歲,因“確診先天性甲狀腺功能減退癥6年,生長緩慢1年”,至中國醫(yī)科大學附屬盛京醫(yī)院小兒內(nèi)分泌科就診。患兒6年前新生兒篩查促甲狀腺激素(thyroid stimulating hormone,TSH)異常,后外院診斷先天性甲狀腺功能減退癥,正規(guī)服用左甲狀腺素鈉治療控制病情,規(guī)律復查甲狀腺功能維持良好。家屬自覺患兒既往偏矮,具體身高增長情況不詳,未特殊診治。1年前患兒開始出現(xiàn)身高增長緩慢,1年僅增長3 cm。患兒平素食量正常,不挑食,無特殊零食及飲料攝入,運動量少。家屬述患兒學習能力尚可,智力正常,溝通能力正常。患兒既往無其他手術外傷史,無傳染病接觸史。患兒系第1胎第1產(chǎn),母孕史、出生史無異常,出生體質量3.2 kg,出生身長50 cm。否認父母近親婚配史,父親身高172 cm,母親身高146 cm,否認佝僂病家族史,否認糖尿病、高血脂及高血壓等家族病史。否認食物及藥物過敏史。
入院體格檢查:體溫36.1 ℃,脈搏88次/min,呼吸20次/min,血壓96/59 mmHg,身高111 cm(位于同年齡同性別第3百分位),體質量23 kg(位于同年齡同性別第3~10百分位),體質量指數(shù)(body mass index,BMI)18.67 kg/m2。神清狀可,體型偏胖,圓臉,五官正常,皮膚無粗糙,無頸璞,雙側乳房TannerⅠ期,無腋毛,心、肺、腹部查體未見明顯異常,雙手第4、5掌骨短,雙足第4、5跖骨短。外生殖器幼稚型,四肢末梢溫,活動自如,神經(jīng)系統(tǒng)查體未見異常。部分臨床特征見圖1,患兒雙手及母親雙手對比見圖2。

圖1 患兒的部分臨床特征
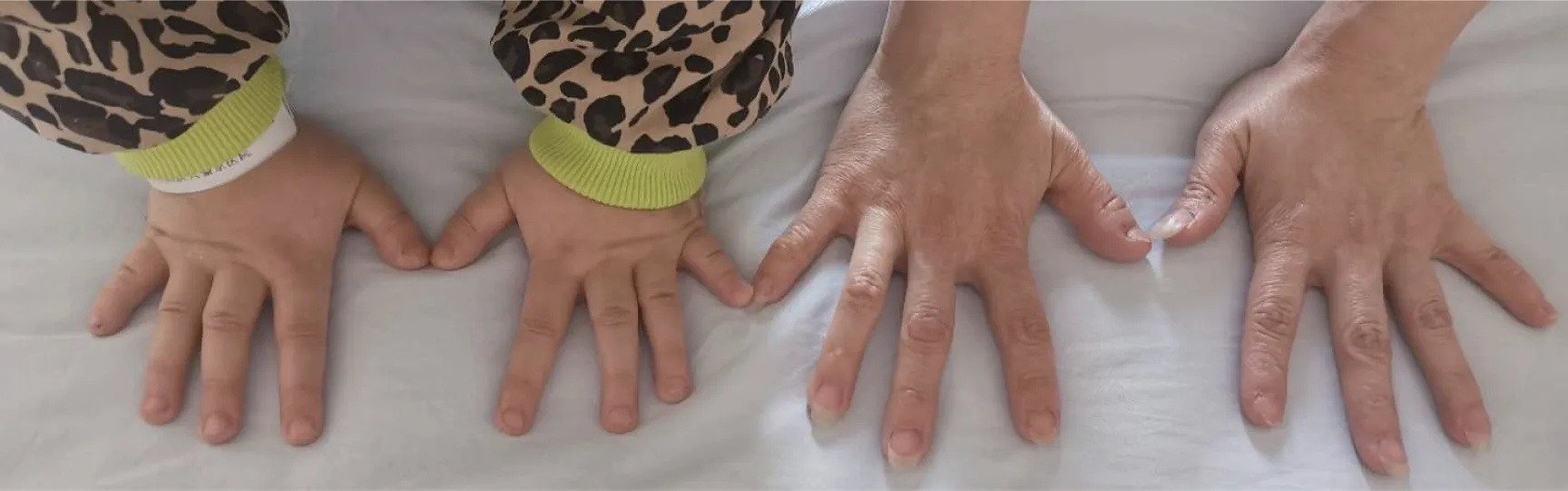
圖2 患兒及母親雙手對比圖
1.2 檢查結果
入院實驗室檢查:血鈣 2.04(2.2~2.7)mmol/L,血磷 2.52(1.2~1.9)mmol/L,甲狀旁腺激素(parathyroid hormone,PTH)499.5(8.0~12.0)pg/mL,24 h尿鈣0.1(2.5~7.5)mmol/d,24 h尿磷 12.95(23.0~48.0)mmol/d,胰島素樣生長因子130(50~410)ng/mL。左手正位X線片:組成諸骨骨質密度不均勻減低,骨小梁稀疏,掌骨及指骨略短縮,部分略彎曲,鉤骨可見囊片狀低密度影,指間關節(jié)間隙變窄,RUS法計算骨齡9歲,見圖3。頭CT:雙側頂葉、基底節(jié)區(qū)見對稱性片狀高密度影,考慮鈣化灶可能性大,其余結構未見明顯異常。
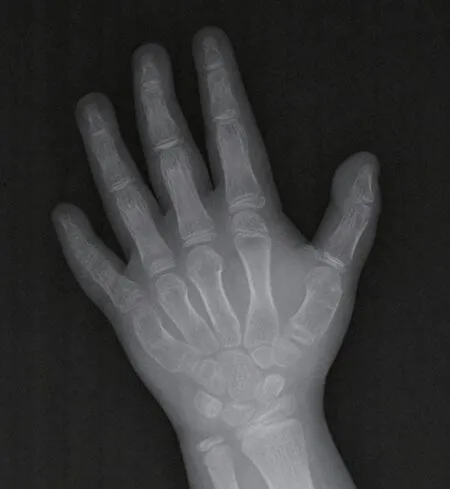
圖3 患兒左手正位X線片
入院后復查甲狀腺功能各指標均在正常范圍內(nèi),血常規(guī)、促腎上腺皮質激素、皮質醇、性激素、人絨毛膜促性腺激素、肝腎功能、心肌酶、血清離子鉀鈉氯檢測,以及心電圖、胸部正側位片、腹腔超聲、雙腎輸尿管超聲、甲狀旁腺超聲、心臟超聲、子宮附件超聲及垂體增強磁共振成像檢查均未見明顯異常。染色體呈46,XX。應用精氨酸及左旋多巴完善生長激素(growth hormone,GH)激發(fā)試驗,測定0、30、60、90、120 min生長激素提示生長激素缺乏,見表1。
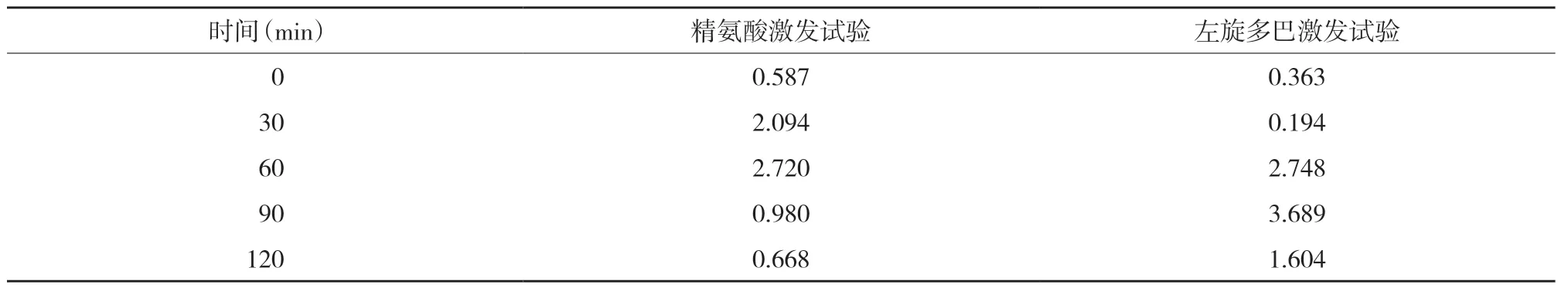
表1 生長激素檢測結果(ng/mL)
1.3 遺傳學分析
結合患兒臨床癥狀、查體及實驗室檢查后,考慮診斷PHP1a型。在獲得知情同意后,抽取患兒及父母靜脈血2 mL,完善基因檢測。按照基因組DNA提取試劑盒(Blood DNA Mini Kit,美國QIAGEN公司)說明書提取DNA,應用超微量分光光度儀(Nanodrop 2000,美國Thermo Fisher Scientific公司)行DNA質檢。合格樣品采用超聲打斷儀(Covaris S220,美國Thermo Fisher Scientific公司)將其打斷至100~700 bp片段,應用標準文庫構建試劑盒(北京邁基諾基因科技有限公司)進行PCR擴增。經(jīng)過生物素標記的探針(北京邁基諾基因科技有限公司)與文庫 DNA 進行雜交,用鏈霉親和素修飾磁珠共價結合生物素標記的探針,抓取目的基因。經(jīng)過質控評估后利用NextSeq 500平臺(德國Illumina公司)完成二代測序分析。利用Cutadapt程序(https://cutadapt.readthedocs.io/en/stable/)[2]過濾掉原始數(shù)據(jù)中低質量的讀取和接頭序列,利用SolexaQA和BWA軟件將預處理后數(shù)據(jù)與人基因組進行比對[3],用GATK程序(https://software.broadinstitute.org/gatk/)對單核苷酸多態(tài)性(single nucleotide polymorphisms,SNPs)和插入/缺失(InDels)進行鑒定,再用ANNOVAR程序(http://annovar.openbioinformatics.org/en/latest/)[4]對識別出的SNPs和InDels進行注釋。最后用MagicViewer軟件(http://bioinformatics.zj.cn/magicviewer/index.php)查看并驗證候選SNPs和InDels。應用4種不同生物信息分析工具Ployphen(http://genetics.bwh.harvard.edu/pph2/)、Sorting Intolerant from Tolerant(SIFT,http://provean.jcvi.org/index.php)、PANTHER(http://www.pantherdb.org)和Pmut(http://mmb.pcb.ub.es/PMut/)對所獲突變進行致病性分析和預測。通過人類基因突變數(shù)據(jù)庫(http://www.biobase-international.com/product/hgmd)支持結果的新穎性,變異點嚴格按照美國醫(yī)學遺傳學和基因組學學院(American College of Medical Genetics and Genomics,ACMG)指南進行致病性分析。
2 結果
遺傳學分析結果顯示,患兒及母親攜帶c.92_94delAGA(P.31_32delQKinsQ)雜合突變,見圖4。該突變?yōu)镚NAS基因轉錄外顯子1號外顯子整碼突變,導致缺失31和32號谷氨酰胺及賴氨酸,插入谷氨酰胺,最終結果為缺失賴氨酸。根據(jù)ACMG指南,該突變?yōu)橐伤浦虏⌒宰儺悾cLOVD數(shù)據(jù)庫(http://www.lovd.nl/GNAS)進行對比,雖該整碼突變?yōu)槭装l(fā),但此位置賴氨酸缺失已有報道[5]與PHP1a型有關,患兒父親未見該位點突變。本例患兒具有先天性甲狀腺功能減退癥、AHO畸形、GH缺乏、甲狀旁腺功能減退癥,并有GNAS基因突變,診斷PHP1a型。二代測序其余結果為無意義突變,故未列舉。出院前予患兒碳酸鈣D3 600 mg口服,3次/d,骨化三醇膠丸0.25 μg口服,2次/d,基因結果回報前暫未應用治療,定期門診復查。
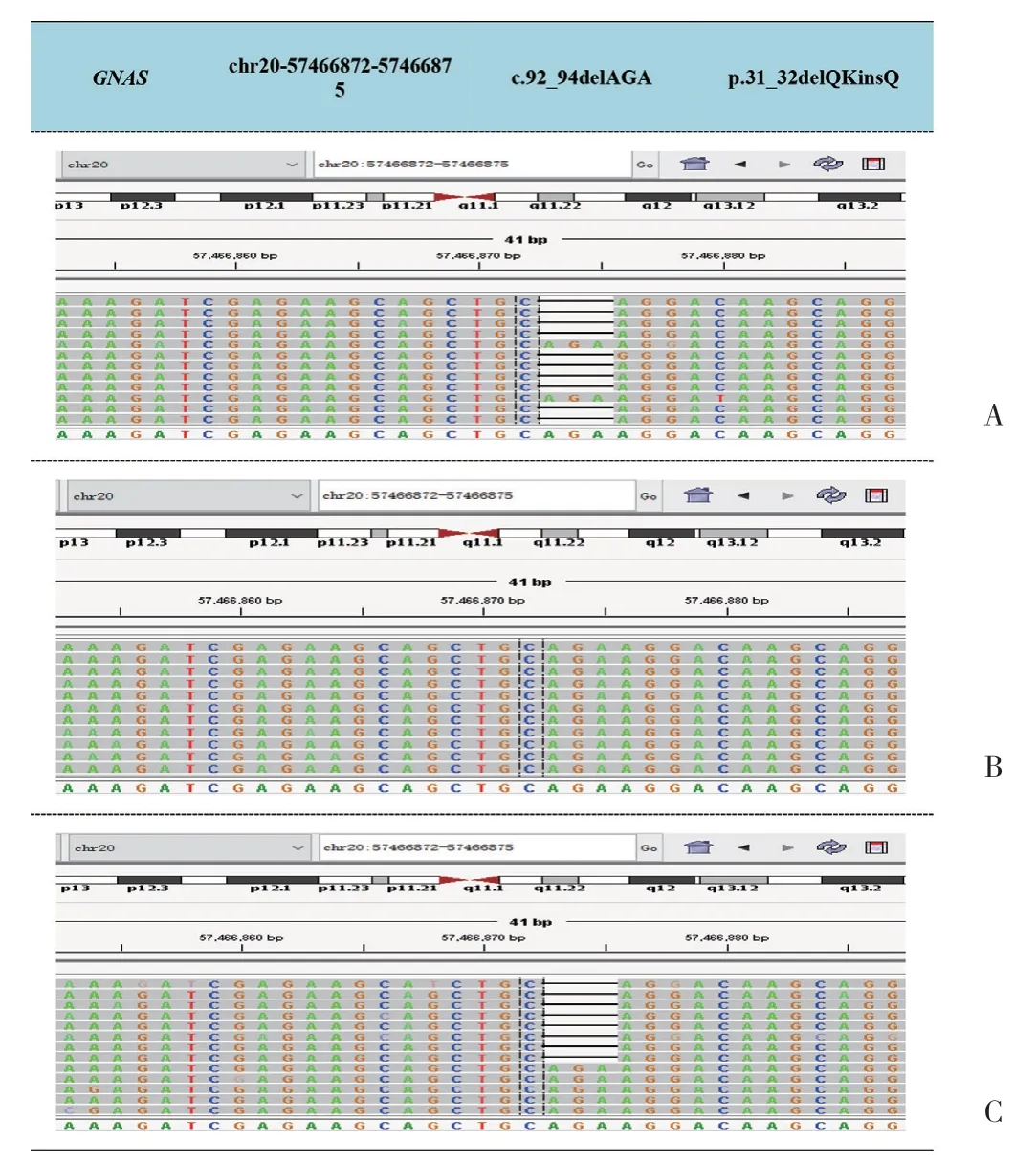
圖4 患兒二代測序基因結果
3 討論
PHP是指由于機體靶組織對PTH產(chǎn)生抵抗所致,具有與甲狀旁腺功能減退癥相同的生化異常(即低血鉀、高血磷)的一組疾病,最早是由ALBRIGHT等[6]首次報道。目前的研究[7]顯示,90%的PHP可發(fā)現(xiàn)分子學證據(jù)。PHP1a型最早被定義為對多種激素(包括甲狀旁腺激素及TSH)抵抗、具有AHO特征改變及與Gsα活性減低相關聯(lián)的疾病[8]。AHO的主要特征包括圓臉、矮胖體態(tài)、脊柱畸形和異位鈣化。身材矮小是由于骨骺過早閉合,骨骼生長發(fā)育期變短所致。骨骼短小可發(fā)生于所有骨骼,但以肢端(即手和腳)短小最為明顯。
目前認為PHP是由6號染色體上的GNAS基因突變造成,遺傳方式為顯性遺傳或新發(fā)突變。本例患兒存在GNAS1號外顯子整碼缺失突變,造成第32號氨基酸缺失賴氨酸,導致Gsα蛋白活性喪失,TSH、PTH及促生長激素抵抗。臨床實驗室檢查可發(fā)現(xiàn)甲狀腺功能減退癥、PHP1a及生長激素缺乏。PHP1a型患者在新生兒期即可發(fā)現(xiàn)TSH抵抗,主要表現(xiàn)為血TSH升高,游離甲狀腺素T4(free thyroxine T4,F(xiàn)T4)正常或輕度降低,可被誤診為先天性甲狀腺功能減退癥[9],國內(nèi)已有文獻[10]報道1例以先天性甲狀腺功能減退癥為首發(fā)癥狀的PHP。陳曉宇等[11]對70例確診先天性甲狀腺功能減退癥患兒進行基因二代測序,發(fā)現(xiàn)3例(4%)存在GNAS基因錯義突變,TSH均>100 μIU/mL,其中2例患兒FT4明顯低于正常值,1例接近正常值下限。PTH抵抗在幼兒期以后才出現(xiàn),并逐漸出現(xiàn)甲狀旁腺功能減退癥樣表現(xiàn),同樣,短指表現(xiàn)至青春期前逐漸明顯[12]。本病例即在新生兒期診斷為先天性甲狀腺功能減退癥,后逐漸因生長緩慢、手指畸形來診并確診。
PHP1a還可能有其他癥狀,如出生身長較正常新生兒短,發(fā)生在其他內(nèi)分泌系統(tǒng)及生化檢查異常之前的肥胖[9]。還有研究[13]發(fā)現(xiàn),PHP1a除了以神經(jīng)系統(tǒng)雙側基底節(jié)鈣化為主的異位鈣化以外,還可能發(fā)生結締組織異位骨化,有異位骨化發(fā)生時不能排除PHP1a。雖然認知障礙與PHP1a有關,但30%的PHP1a患者認知發(fā)育正常[14]。除上述癥狀外,Gsα蛋白活性喪失還可能造成降鈣素、促黃體生成素及促卵泡生成素抵抗,導致這些激素水平升高。
臨床高度懷疑PHP的患者最終需通過分子診斷確診。如前所述,PHP1a型主要是由于GNAS基因位點突變所致,突變類型可以是母系遺傳也可為新發(fā)。1代測序及2代測序均可發(fā)現(xiàn)基因點突變,而基因重組突變或基因甲基化缺陷需通過多重連接探針擴增技術發(fā)現(xiàn)[15]。尚無證據(jù)表明GNAS突變的類型與疾病發(fā)病時間、內(nèi)分泌激素抵抗的嚴重程度、神經(jīng)認知表型或AHO特征的數(shù)量之間具有相關性。
目前,對PHP以基于患者癥狀及實驗室檢查異常的治療為主。甲狀腺旁腺激素抵抗需補充活性維生素D或類似物及鈣劑,以抑制PTH水平過高及低鈣血癥、高磷血癥。前者會導致骨骺過早鈣化,后者則可致腎及顱內(nèi)鈣沉積。TSH抵抗治療原則與甲狀腺功能減退癥或亞臨床甲狀腺功能減退癥治療原則相同,但是對甲狀腺功能減退癥的及時診斷與治療似乎不能阻止PHP1a型患者運動或認知障礙的發(fā)展。幾乎所有的PHP1a型患者均有身材矮小癥狀,因此,對于身材矮小合并骨骼畸形的患者,應額外注意PHP的可能。PHP1a所導致的肥胖及其他激素的抵抗也應長期關注隨訪。
PHP1a型患者從出生時即可發(fā)生甲狀腺功能減退癥,之后出現(xiàn)身材矮小,假性甲狀旁腺功能亢進所致骨骺過早閉合、異位骨化及異位鈣沉積所致驚厥發(fā)作,骨骼畸形,活動受限,認知障礙和精神異常。及早發(fā)現(xiàn)和治療可減輕或延后相關并發(fā)癥的進展。
猜你喜歡
鐘表(2023年5期)2023-10-27 04:20:44
中華詩詞(2022年6期)2022-12-31 06:41:24
當代陜西(2021年21期)2022-01-19 02:00:26
中學生數(shù)理化(高中版.高考數(shù)學)(2020年1期)2020-02-20 13:23:44
經(jīng)濟技術協(xié)作信息(2018年11期)2019-01-14 03:07:20
中國科技論壇(2017年7期)2017-07-25 08:49:53
制造技術與機床(2017年3期)2017-06-23 08:11:33
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
中國中醫(yī)藥現(xiàn)代遠程教育(2014年22期)2014-03-01 04:32:55
