超高效液相色譜-串聯質譜法測定配制酒中甜蜜素的含量
2023-07-24 00:57:16張嘯怡何青科徐文泱
食品安全導刊 2023年19期
關鍵詞:方法
劉 慧,張嘯怡,何青科,徐文泱
(湖南省產商品質量檢驗研究院,食品安全監測與預警湖南省重點實驗室,湖南長沙 410017)
甜蜜素(環己基氨基磺酸鈉)是一種以環己胺為原料,經氯磺酸或氨基磺酸化形成環己基氨基酸磺酸后與氫氧化鈉作用而制成的人工合成甜味劑,其甜度是蔗糖的30 ~80 倍,常在配制酒、冷凍飲品、腌漬蔬菜、餅干、飲料、糕點、蜜餞和復合調味料等食品中添加使用。人體不吸收甜蜜素,幾乎全部以原型經糞便排出,在體內不蓄積[1]。
配制酒通常以發酵酒或蒸餾酒及食用酒精為酒基,加入可食用動植物組織或既是食品又是中藥材的物質,輔加食品添加劑用于呈色呈味。由于甜蜜素具有較好的改善配制酒風味和口感的作用,部分配制酒生產者在實際生產過程中通常會添加一定量的甜蜜素以調整配制酒的適口性。《食品安全國家標準 食品添加劑使用標準》(GB 2760—2014)[2]規定,配制酒中甜蜜素的最大使用量為0.65 g·kg-1(以環己基氨基磺酸計)。臨床實踐表明,長期或過量攝入甜蜜素,尤其是此類合成甜味劑,會對人體的代謝系統、神經系統等造成影響[3-4]。聯合國糧農組織和世界衛生組織食品添加劑聯合專家委員會建議其每日容許攝入量為0 ~11 mg·(kg bw)-1。
目前,測定食品中甜蜜素的檢測方法主要包括分光光度法[5]、液相色譜法(LC)[6]、氣相色譜法[7-8]、離子色譜法[9]、氣相色譜質譜聯用法[10-11]和液相色譜質譜聯用法[12-14]。對于配制酒中甜蜜素測定的現行有效的食品安全國家標準方法為GB 5009.97—2016 第二法 高效液相色譜法[15],該方法通過衍生化將環己基氨基磺酸鈉水提后,在強酸性溶液中與次氯酸鈉反應,生成N,N-二氯環己胺,經正庚烷萃取,利用高效液相色譜法檢測,操作煩瑣,定量限為0.030 g·kg-1,易出現“假陰性”結果。
由于超高效液相色譜-串聯質譜法具有快速分離、靈敏度高、選擇性高的優勢,已廣泛應用于白酒、飲料、糕點和面制品中甜蜜素的測定。本文以配制酒和甜蜜素為研究對象和目標,開展相關方法學考察,建立配制酒中甜蜜素的測定方法。通過實際樣品測定,比較本方法和GB 5009.97—2016 高效液相色譜法二者測定結果的差異,為實現開發配制酒中甜蜜素簡便、快速、準確、靈敏的液質聯用檢測方法提供研究基礎,為打擊配制酒中甜蜜素超量使用等違法違規行為提供檢驗依據,以滿足食品安全監管需求。
1 材料與方法
1.1 材料與儀器
配制酒樣品23 批次,來源于湖南省流通和生產領域的監督抽查;環己基氨基磺酸鈉標準品,C6H11NHSO3Na,CAS:139-05-9,純度≥99.9%,0.25 g,DR;甲醇,色譜純,上海安譜實驗科技股份有限公司;乙酸銨,色譜純,阿拉丁生化科技股份有限公司;超純水采用Milli-Q 制備(18.2 MΩ·cm)。
Thermoscientific Vanquish+TSQ QUANTIS 超高效液相色譜質譜聯用儀;Sartorius SECURA225D-1CN 分析天平。
1.2 溶液的配制
(1)標準儲備液。準確稱取11.2 mg 環己基氨基磺酸鈉用水溶解定容至10 mL,搖勻,制成濃度為1.0 mg·mL-1的標準溶液。
(2)標準中間液。吸取上述標準儲備液適量,用水溶解定容至10 mL,搖勻,制成濃度為1.0 μg·mL-1的標準中間液。
(3)標準曲線系列工作液。吸取上述標準中間液適量,用水逐級稀釋制成5 ng·mL-1、10 ng·mL-1、20 ng·mL-1、50 ng·mL-1、100 ng·mL-1、200 ng·mL-1和500 ng·mL-1標準系列溶液。
1.3 樣品前處理
稱取樣品1.0 g 置于100 mL 比色管中,加水稀釋至刻度,搖勻,過0.22 μm 濾膜,待測。
1.4 儀器條件
1.4.1 液相色譜條件
色譜柱:Hypersil GOLD C18(150 mm×2.1 mm,3 μm); 柱溫:30 ℃ ; 進樣量:5 μL; 流速:0.2 mL·min-1;流動相:10 mmol·L-1乙酸銨(A)和甲醇(B),梯度洗脫條件見表1。
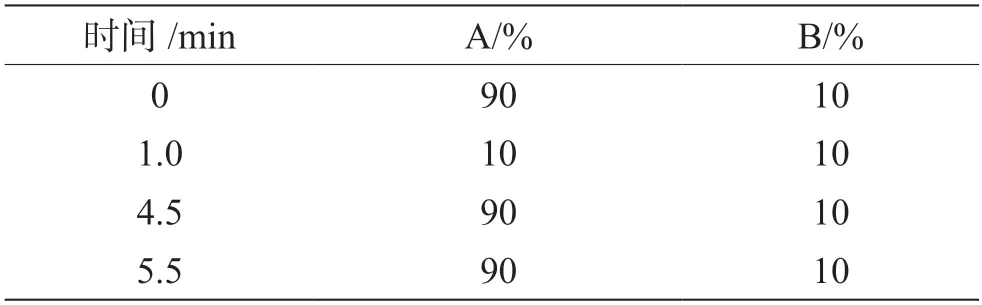
表1 流動相梯度洗脫條件
1.4.2 質譜條件掃描方式:正離子掃描(ESI-);毛細管電壓:2 500 V;鞘氣流速:20 Arb;輔助氣流速:5 Arb;離子傳輸管溫度:325 ℃;離子源溫度:350 ℃;掃描方式:多反應離子監測(MRM),條件見表2。

表2 甜蜜素多反應離子監測條件
2 結果與分析
2.1 試驗條件優化
2.1.1 樣品前處理
配制酒的酒基為蒸餾酒及食用酒精或發酵酒,因此配制酒中含有一定濃度的乙醇。根據白酒中甜蜜素分析方法前處理過程需于60 ℃水浴加熱30 min去除酒精,考慮配制酒中乙醇含量對測定結果的影響,本試驗比較揮發樣品中乙醇(60 ℃水浴上加熱30 min)與直接用水稀釋兩種前處理方式對甜蜜素測定結果的影響,結果見表3。3 個濃度水平兩種前處理方式的測定結果相對偏差為-12.2%~5.0%,無明顯差別,可能是因為配制酒不同于高乙醇含量的白酒,其乙醇濃度偏低,用水稀釋后,乙醇對測定結果的影響可忽略不計。直接稀釋處理進樣快速、簡便,因此,本試驗選擇直接用水稀釋進樣分析。
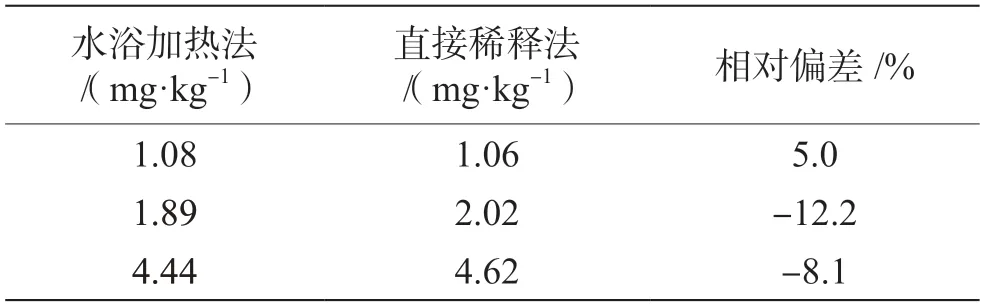
表3 兩種前處理方法甜蜜素含量測定結果
2.1.2 色譜與質譜條件
由于甜蜜素(環己基氨基磺酸鈉)以鈉鹽形式存在,在水溶液中易電離形成[M-Na]-,適合負離子模式進行檢測。ESI 離子化過程主要受化合物濃度、表面活性、基質(溶劑)性質、pH 值以及流速的影響,在流動相中添加適當的改性劑如甲酸、氨水、乙酸銨,有助于提高化合物離子化效率和改善峰型。負離子模式下,乙酸銨、氨水更容易奪取溶劑中的H+,從而促使甜蜜素從分子態向離子態的轉化,提高電離效率。但考慮到氨水對色譜柱的損傷,本試驗采用乙酸銨作為改性劑,通過比較5 mmol·L-1、10 mmol·L-1、15 mmol·L-1乙酸銨溶液對響應和峰形的影響發現,在10 mmol·L-1乙酸銨甜蜜素響應好且峰形佳,因此最終采用10 mmol·L-1乙酸銨水溶液作為流動相。
2.2 方法學驗證
2.2.1 專屬性
按上述過程制備配制酒樣品、配制酒樣品中加入標準溶液的基質加標樣品,吸取空白溶劑、標準溶液、樣品溶液和基質加標溶液進樣分析,色譜圖見圖1。樣品溶液與基質加標溶液色譜在甜蜜素峰位有相同的色譜峰出現,而空白溶液在相應位置上無其他干擾峰,方法專屬性良好。
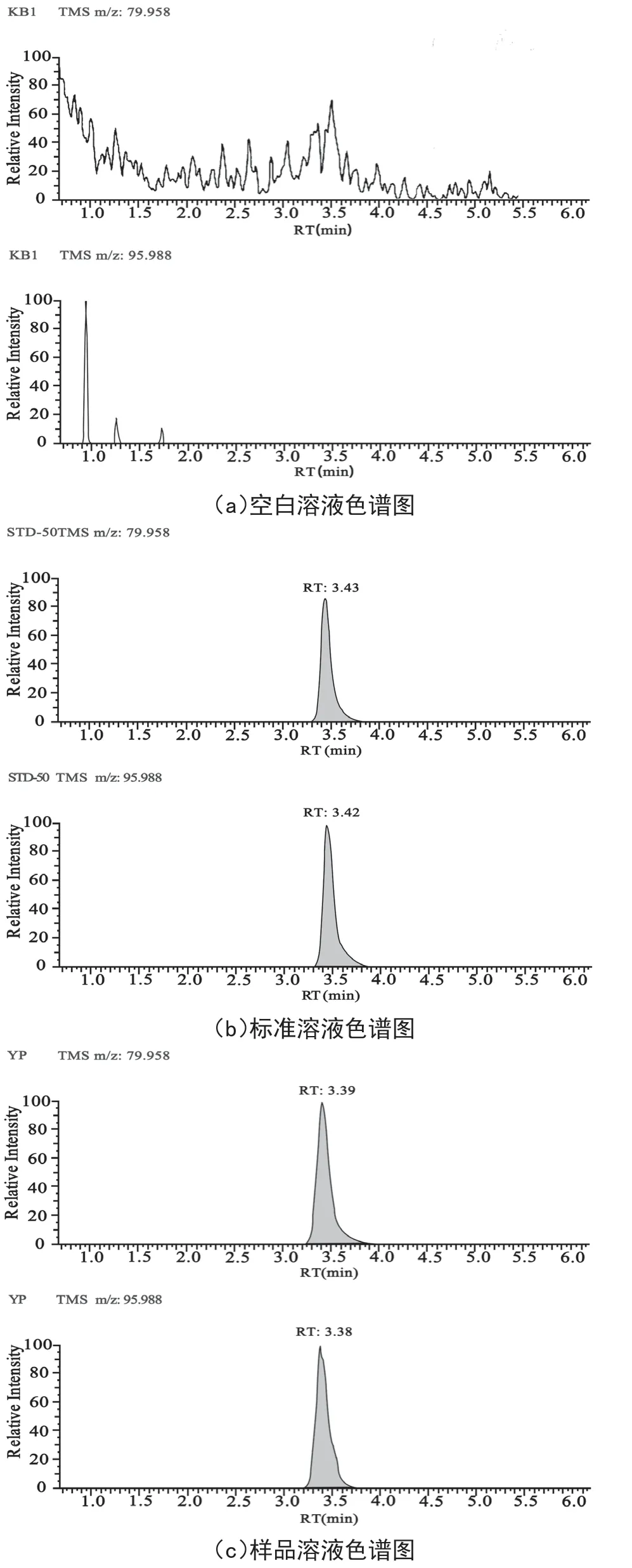
2.2.2 標準曲線
取1.2 項下標準曲線系列工作液,按上述條件測定,以甜蜜素峰面積為縱坐標、濃度為橫坐標繪制標準曲線,得出甜蜜素線性方程為y=7.886×103x-2.631×104(r2=0.999 1),甜蜜素在5 ~500 ng·mL-1線性關系良好。
2.2.3 檢出限和定量限
將標準溶液稀釋至適當濃度,按1.3 項下制備適當濃度的基質加標樣品溶液,以S/N=3 和S/N=10 分別計算儀器和方法的檢出限和定量限,甜蜜素的儀器檢出限和定量限分別為0.15 ng·mL-1和0.3 ng·mL-1,方法檢出限和定量限分別為0.01 mg·kg-1和0.05 mg·kg-1。
2.2.4 準確度試驗
取配制酒樣品(甜蜜素含量為13.5 mg·kg-1),在樣品中添加低、中、高3 個水平濃度的甜蜜素標準溶液,每個水平6 份,按上述方法進行處理,計算甜蜜素回收率和相對標準偏差,結果見表4。甜蜜素在2.5 ~10.0 mg·kg-1添加水平下,回收率為99.0%~100.6%,相對標準偏差為0.8%~1.6%,表明該方法準確可靠,可滿足分析方法要求。

表4 甜蜜素回收率試驗結果
2.2.5 精密度試驗
取配制酒陽性樣品(甜蜜素含量為55.6 mg·kg-1),按1.3 項處理,重復測定6 次,測定結果分別為57.6 mg·kg-1、56.9 mg·kg-1、57.0 mg·kg-1、55.6 mg·kg-1、56.5 mg·kg-1和55.5 mg·kg-1,相對平均偏差為1.5%,相對誤差為1.6%,表明方法重復性良好。
2.2.6 稀釋穩定性
取配制酒樣品,添加甜蜜素標準溶液制成甜蜜素含量為100 mg·kg-1的樣品溶液,按前處理過程進行處理,將處理的樣品溶液分別稀釋10 倍和100 倍,每個稀釋度6 份,測定,計算相對平均偏差和相對誤差,稀釋10倍和100倍的相對平均偏差分別為2.6%和3.2%;相對誤差分別為-0.3%和-1.3%,表明超線性范圍濃度的樣品,可通過對樣品進行一定程度的稀釋,此稀釋過程穩定,不影響結果的準確性。
2.3 樣品測定
將流通領域抽查的23 份配制酒樣品按已建立的方法進行測定分析,并與GB 5009.97—2016 第二法測定結果進行比較,結果見表5。
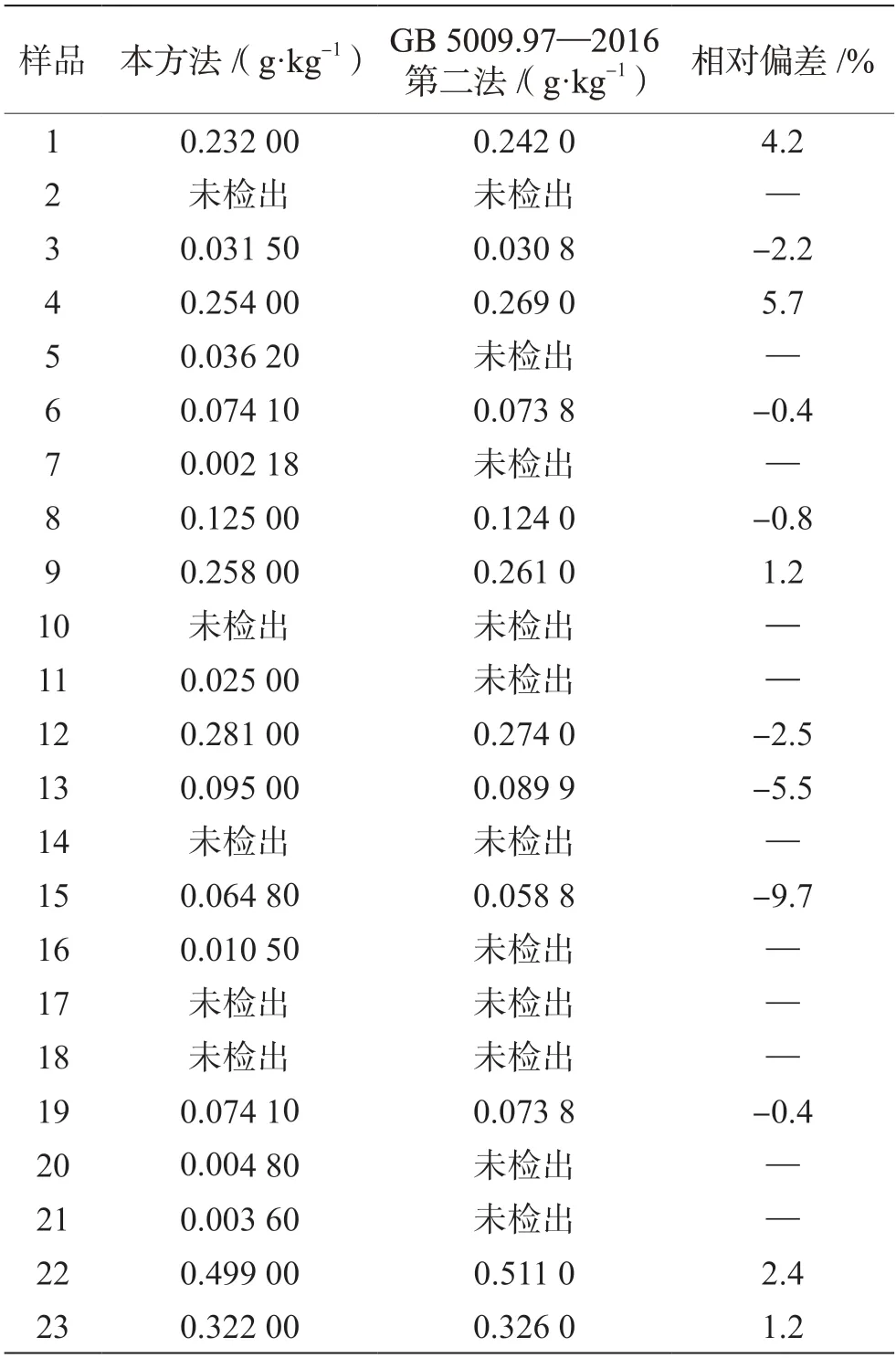
表5 本方法與GB 5009.97—2016 第二法關于23 批配制酒中甜蜜素含量測定結果的比較
本方法共18份樣品檢出甜蜜素,含量為0.002 18~0.499 00 g·kg-1;GB 5009.97—2016 第二法共12 份樣品檢出甜蜜素,含量為0.030 8 ~0.511 0 g·kg-1。6 份樣品本方法有檢出,GB 5009.97—2016 第二法未檢出;5 份樣品兩種方法均未檢出,12 份樣品兩種方法均檢出,相對偏差為-9.7%~5.7%。所測定的配制酒樣品中甜蜜素含量結果均≤0.65 g·kg-1(以環己基氨基磺酸計),符合GB 2760—2014 使用限量要求。
3 結論
本文建立了配制酒中甜蜜素含量的超高效液相色譜-串聯質譜測定方法,并開展方法學考察以驗證方法的可靠性。通過實際樣品測定,比較本方法和GB 5009.97—2016 高效液相色譜法二者測定結果的差異,對于均檢出甜蜜素的樣品兩種方法結果相對偏差<15%,部分高效液相色譜法未檢出的樣品,本方法有檢出,在一定程度可避免“假陰性”結果。該方法簡便、快速、準確、靈敏,可為打擊配制酒中甜蜜素超量使用等違法違規行為提供檢驗依據,以滿足食品安全監管需求。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
