家族性噬血細胞性淋巴組織細胞增生癥合并UNC13D基因突變3例病例總結并文獻復習
2023-07-28 08:49:58錢冰濤王澤芯羅榮牡
罕少疾病雜志 2023年7期
關鍵詞:基因突變
錢冰濤 王 萌 王澤芯 羅榮牡 宋 慶
航天中心醫院兒科 (北京 海淀 100049)
噬血細胞性淋巴組織細胞增生癥(h e m o p h a go c y t i c lymphohistiocytosis,HLH)又稱為噬血細胞綜合征,可以累及任何年齡的個體,最常見于嬰兒和年幼兒童,是一種危重的侵襲性免疫過度活化綜合征。HLH臨床表現上以持續性發熱、肝脾腫大、全血細胞減少,多器官功能損害甚至衰竭,其病因是由于巨噬細胞、自然殺傷(NK)細胞、細胞毒T淋巴細胞等異常活化引起的全身過度炎癥反應綜合征[1]。本病罕見,臨床表現不典型、復雜多樣,且其病情進展迅速,病情危重,治療難度大,死亡率高,診斷明確后應當及時治療。近些年隨著醫療技術發展以及對HLH的病因研究,各級醫院臨床醫生對HLH的認識越來越深入,早發現,早診斷,早治療,使得HLH臨床診治水平提高。
噬血細胞性淋巴組織細胞增生癥(HLH)原發性和繼發性兩大類[2]。原發性HLH包括家族性HLH(FHL)及遺傳性免疫缺陷綜合征,由基因突變導致。目前為止已確認的FHL有5種類型:FHL1、FHL2、FHL3、FHL4、FHL5。其中4種FHL均已明確其致病基因,分別為PRF1、UNC13D、STX11、STXBP2。遺傳性免疫缺陷綜合征患兒往往具有免疫缺陷、生長發育異常等表現,包括Chediak-Higash綜合征、Griscelli綜合征、X連鎖淋巴組織增生癥、Wiskott-Aldrich綜合征、嚴重的聯合免疫缺陷病[3]。原發性HLH發病年齡早,多在嬰幼兒期發病,故原發性HLH多發于兒童,成年人原發性HLH較為少見。
臨床上最常見的是繼發性HLH,可繼發于感染性疾病、腫瘤、自身免疫性疾病、造血干細胞移植及藥物過敏等。感染相關性HLH其病因感染源可以是病毒、細菌、真菌或寄生蟲,臨床上EB病毒感染導致的相關性噬血細胞綜合征(EBV-HLH)最為常見[4]。兒童持續性EBV感染,可能會發展為慢性活動性EBV感染,從而導致繼發性HLH[5-6]。
HLH在發病過程中存在巨噬細胞、NK細胞和CTLs持續活化,以至于產生過量細胞因子(細胞因子風暴),導致多臟器功能受損、多器官衰竭,這是HLH死亡率高的原因[7]。未經醫院綜合治療的HLH自然病程幾乎都是致命的。本病罕見且臨床表現復雜,病因多樣,故而及時識別HLH,并診斷明確HLH的病因,對于盡早行綜合治療改善預后至關重要。近些年基因檢測技術飛速發展,HLH患兒診斷后進行基因檢測,越來越多的家族性HLH(FHL)浮出水面,討論原發性HLH患兒基因型不同,其臨床表型是夠有特異性。原發性HLH盡早評估病情,化療后行造血干細胞移植(hematopoietic cell transplant, HCT),有助于改善患兒預后。本研究對3例家族性HLH(FHL)臨床資料進行了收集,分析患兒的臨床特點及實驗室、功能學檢查結果的差異,為HLH患兒的個體化治療提供一定依據。
1 資料與方法
1.1 一般資料資料選取2020年5月至2022年10月北京航天中心醫院兒科住院并確診為家族性HLH(FHL)臨床資料進行回顧性分析,均通過航天中心醫院醫學倫理委員會批準,且監護人簽署知情同意書。隨訪:3例FHL患兒來院門診復查或住院治療進行隨訪。
納入標準:3例均符合中國專家共識(2018年版)中噬血細胞綜合征診斷標準[8]。(2)3例患兒均行噬血細胞綜合征相關基因檢測。HLH累及中樞神經系統(CNS-HLH)診斷標準國際目前沒有統一定論[9]。很多專家認為中樞神經系統受累指的是HLH患者出現一種或多種中樞神經系統和/或精神癥狀、神經影像學、腦脊液的異常,并且排除其他因素。CNS-HLH可以分為三個病理學階段:Ⅰ期:活化的淋巴細胞、巨噬細胞只浸潤軟腦膜;Ⅱ期:浸潤周圍血管間隙;Ⅲ期:腦實質的廣泛浸潤,引起血管破壞及腦組織的廣泛多灶性壞死、脫髓鞘病變,最后出現神經元丟失和膠質增生[2]。
1.2 方法回顧性分析3例家族性噬血細胞性淋巴組織細胞增生癥(FHL)患兒的臨床資料,從病歷中收集并分析患兒的臨床表現,常規實驗室檢查結果,EB病毒抗體及血清EBV-DNA檢測結果,T、B、NK細胞檢測結果,影像學檢查報告,遺傳學測序結果(原發性HLH相關基因篩查)以及自然殺傷(NK)細胞活性、可溶性CD25分子(sCD25正常值<6400ng/L)。
2 結 果
2.1 一般資料收集3例家族性噬血細胞綜合征FHL,3例患兒UNC13D基因突變均陽性,均行化療后進行造血干細胞移植(hematopoietic cell transplant, HCT)。
2.2 臨床癥狀及體征病例1,5歲,女性,以“皮疹、發熱、淋巴結腫大”起病,病初頭面部、全身出現丘疹樣皮疹,高出皮面,部分破潰、滲出,間斷發熱,體溫最高達39℃,熱峰1次日,流黃涕,鼻孔粘膜增生,查體耳后、頸部淋巴結腫大,腹部觸診肝脾腫大。皮疹好轉后,患兒發熱、抽搐1次,查頭顱核磁MRI提示胼胝體壓部有異常信號,考慮伴胼胝體壓部存在可逆性病變、臨床癥狀輕微的腦炎腦病(Mers)。小腦腦溝稍寬。左頂頭皮下異常信號,考慮皮樣囊腫可能。EB病毒抗體檢測:EB病毒衣殼抗原(CA)IgG陽性;EB病毒核抗原(NA)抗體陽性。淋巴結病理示:右頜下淋巴結EB病毒陽性T細胞增殖性疾病(Ⅱ級)。復查頭顱核磁提示腦室系統稍擴大,腦溝腦裂略增寬,輕度腦白質發育異常,輕度鼻竇炎。給予化療及對癥治療,并行造血干細胞移植術,定期隨訪。

表1 噬血細胞綜合征臨床特點
病例2,3歲,男性,以“腹痛、腹脹”起病,之后出現發熱,查體頸部淋巴結腫大,肝脾腫大,完善骨髓穿刺術送檢未見噬血細胞現象,EB病毒DNA測定1.09×104copies/mL,抗病毒治療無效,患兒間斷發熱,出現皮疹,該患兒病史、體征以及實驗室檢查,臨床診斷HLH,復查骨穿可見噬血細胞現象,診斷噬血細胞性淋巴組織細胞增生癥(HLH),給予化療及對癥治療,并行造血干細胞移植術,定期隨訪。
病例3,6月,男性,以“發熱、皮疹”起病,病初高熱,體溫39℃,熱峰4次/日,散在紅色皮疹,抗感染治療效果不佳,持續高熱,根據患兒臨床表現、體征以及實驗室檢查診斷噬血細胞性淋巴組織細胞增生癥(HLH),骨髓穿刺術送檢骨髓可見噬血細胞現象,給予化療及對癥治療,并行造血干細胞移植術,定期隨訪,生長發育落后。
2.3 臨床實驗室檢查資料整理
2.4 影像學檢查資料
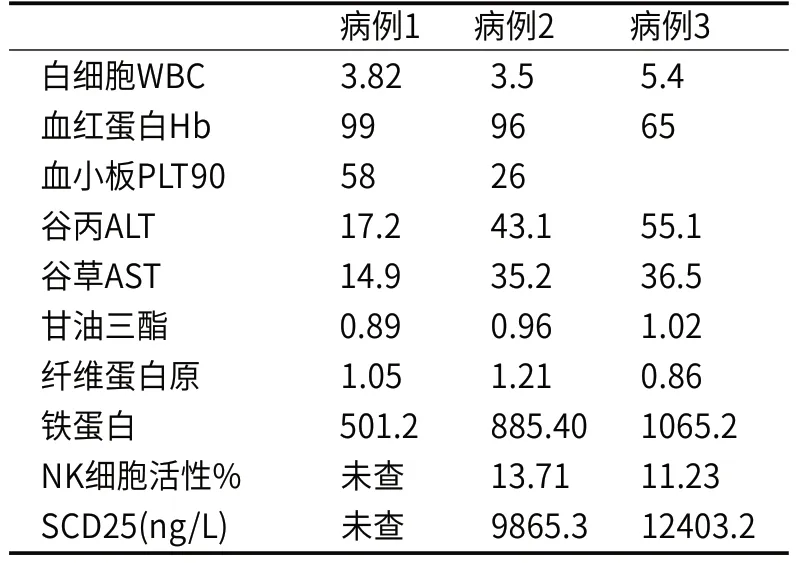
表2 臨床實驗室檢查結果
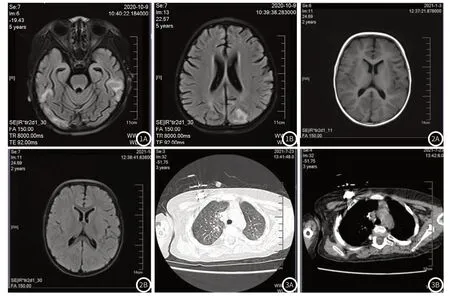
病例1 5歲,有發熱、抽搐癥狀,頭顱核磁提示雙側側腦室周圍白質內可見暈狀T1WI等信號、T2WI高信號、FLAIR高信號灶。DWI未見異常高信號。腦室系統稍擴大,腦溝腦裂略增寬。輕度腦白質發育異常。(見圖1A-圖1B)病例2 6月發病,生長發育落后,頭顱核磁:左側放射冠可見點狀T2WI及FLAIR高信號,左側放射冠點狀異常信號灶,脫髓鞘。DWI未見明顯高信號。左側顳部蛛網膜囊腫。(見圖2A-圖2B)病例3 6月發病,反復感染性肺炎,出現并發癥閉塞性支氣管炎,胸部CT提示支氣管管壁增厚,雙肺多發淡片狀磨玻璃影,雙肺下葉呈馬賽克改變,右肺尖胸膜下索條影,右肺中葉、左肺下舌段條片狀實變影。(見圖3A-圖3B)
3 討 論
噬血細胞性淋巴組織細胞增生癥(HLH)發病機制中涉及的細胞類型包括2類:1.巨噬細胞是起源于循環中單核細胞的專職抗原提呈細胞,可將外源性抗原提呈給淋巴細胞。HLH巨噬細胞出現異常活化,并分泌過量的細胞因子,最終導致組織重度損傷,可引起器官功能衰竭。2.自然殺傷細胞(natural killer cell)占到淋巴細胞數的10%-15%。NK細胞通常可以清除受損或被感染的宿主細胞(例如巨噬細胞)。HLH患兒體內NK細胞和/或CTLs不能夠清除那些異常活化的巨噬細胞。由于缺失以上正常反饋調節,CD8+ T細胞和巨噬細胞出現過度活化,IFN-γ、細胞因子水平異常升高,從而導致HLH的病理變化[10-11]。HLH患兒體內免疫系統Toll樣受體(tolllike receptor, TLR)活化可能是導致HLH的另一個原因[12]。
HLH其他免疫紊亂,比如淋巴細胞異常包括CD4和CD8淋巴細胞亞群數量改變。2015年納入21例HLH患者中有20例出現淋巴細胞亞群和/或免疫表型異常。其中10例出現CD8+細胞增多和CD4:CD8比率降低,8例出現CD3+細胞減少,3例出現CD56+細胞增多、CD7-/CD4+細胞增多和CD4+細胞增多。病例系列研究顯示:CD8數量增加和CD4/CD8比值下降的患者生存情況最佳。CD3總數下降預示著患者預后較差[13]。
收集到的3例原發性家族性HLH均為UNC13D基因突變陽性。在原發性HLH攜帶破壞性基因突變的患兒發病年齡較早;家族性HLH(FHL)的患兒較繼發性HLH更容易出現中樞神經系統受累,出現抽搐、嗜睡、精神異常等;FHL脫顆粒通路受損患兒sCD25、膽紅素及IFN-γ、IL-10水平較高[14]。Unc(即uncoordinated)蛋白家族調節溶細胞顆粒的成熟,UNC13D/Munc13-4-編碼Munc13-4的UNC13D基因突變會導致FHL3[15]。
HLH除了單個HLH基因的純合性突變外,HLH也可能為復合雜合子突變(同一基因的兩個等位基因上可能存在不同的突變)或為雙基因遺傳(在兩個不同的基因上可能存在不同的突變)。一項回顧性研究納入了2701例HLH就診接受基因檢測的患者,結果發現225例(8%)患者為純合性突變或復合雜合型突變,其中28例(1%)患者為雙基因遺傳,有21例在PRF1和脫顆粒基因內發生突變,7例在參與脫顆粒途徑的2個基因內發生突變。此項大樣本研究表明,由于參與細胞毒性淋巴細胞脫顆粒的基因內的協同功能效應,FHL具有潛在的雙基因遺傳模式[16]。
另一項研究報道了類似結果,納入了281例歸為“散發性”HLH的患者中,43例存在已知的家族性HLH基因的單等位基因突變,提示該病并非單純通過隱性遺傳。基因SPRF1(FHL2)和UNC13D(FHL3)的突變占FHL病例的70%。因此90%以上的家族性HLH(FHL)患者可以進行基因診斷。穿孔素表達和脫顆粒程度對FHL的診斷比噬血細胞和細胞毒性試驗更有用。FHL并不是嚴格的隱性遺傳。專家們認為臨床綜合征HLH通常是由外源性觸發因素和遺傳易感性共同作用的結果。外源和遺傳因素的不同權重解釋了從HLH繼發到嚴重感染到FHL的廣泛疾病譜[17]。并且盡早進行基因檢查,有助于明確是否為FHL,有助于制定治療計劃,盡早行骨髓移植治療。
一篇文章報道了1例患兒,主要表現為發熱,伴腹脹、鼻衄,當地醫院診斷慢性活動性EB病毒感染,伴有肝功能異常、肝臟纖維化,入院后診斷為繼發性HLH,住院對癥治療好轉后出院,1月后隨訪患兒病情惡化,因多臟器功能衰竭死亡[18]。因此本病容易出現多臟器受累,病情進展迅速,對癥治療及化療治療效果不理想,預后極差。家族性HLH(FHL)攜帶基因突變,病情更為兇險,應盡早完善相關檢查評估病情,盡早行全面治療。
綜上所述,家族性HLH(FHL)發病年齡較小的患兒遺傳信息有助于評估其HLH復發風險、造血干細胞移植的必要性,以及家庭成員出現HLH的風險。HLH發病年齡越小,發現基因突變概率越高,因此對于小年齡起病的反復發熱、皮疹、肝臟脾臟腫大患兒,診斷為噬血細胞綜合征建議盡早進行基因突變的篩查。基因篩查有異常,確診為家族性HLH(FHL)病情進展迅速,化療及對癥治療后,建議盡早行造血干細胞移植有助于改善預后。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
