瑞馬唑侖對腦缺血再灌注損傷模型大鼠腦神經的保護作用及其機制
2023-08-01 08:30:54段梅劉佳張靜孫曉麗王士雷
精準醫學雜志 2023年4期
關鍵詞:血清
段梅 劉佳 張靜 孫曉麗 王士雷
(青島大學附屬醫院麻醉科,山東 青島 266555)
缺血性腦卒中是全球人群殘疾和死亡的主要原因之一[1],早期恢復缺血性腦卒中患者腦血流供應是其最有效治療方法[2]。然而血流恢復過程會導致進一步的再灌注損傷,稱之為腦缺血再灌注(I/R)損傷[3]。核因子κ-活化B細胞輕鏈增強子(NF-κB)是一種關鍵的核轉錄因子,在腦I/R損傷中被激活,通過增加促炎細胞因子白細胞介素-6(IL-6)、IL-1β以及腫瘤壞死因子-α(TNF-α)的產生而放大炎癥反應[4]。NF-κB p65是NF-κB家族的主要成員[5],其磷酸化以及核易位是NF-κB途徑激活的最直接的證據[6]。蛋白激酶B(PKB,也稱AKT)是磷脂酰肌醇3-激酶(PI3K)的主要的下游靶標,磷酸化的AKT(p-AKT)是AKT的活性形式。研究表明,甘氨酸通過激活AKT抑制NF-κB p65/Hif-1α信號通路,從而減輕腦缺血誘導的炎癥反應[7]。瑞馬唑侖是一種新型苯二氮類γ-氨基丁酸A受體激動劑,適用于誘導和維持成人的程序性鎮靜。研究表明在神經性疼痛的發作過程中,瑞馬唑侖可以抑制NF-κB核易位,并減少促炎細胞因子的表達[8]。此外,瑞馬唑侖可抑制NOD樣受體熱蛋白結構域相關蛋白3炎性小體介導的炎癥級聯反應,減少神經元損傷,從而減輕腦I/R損傷[9]。然而,瑞馬唑侖對腦I/R損傷的保護機制十分復雜且有待深入研究。本研究旨在觀察瑞馬唑侖對腦I/R損傷的保護作用,并探究AKT/NF-κB信號通路是否參與該過程。現將結果報告如下。
1 材料和方法
1.1 材料與試劑
雄性SD大鼠(體質量230~260 g)購自濟南朋悅實驗動物繁育有限公司,PI3K抑制劑LY294002購買于美國MedChemExpress公司,AKT、p-AKT、NF-κB p65及磷酸化NF-κB p65(p-NF-κB p65)抗體均購買于美國Cell Signaling Technology公司,IL-6、IL-1β和TNF-α酶聯免疫吸附試驗(ELISA)試劑盒均購自上海勁馬生物科技有限公司。
1.2 分組及處理
所有大鼠在恒溫25 ℃室內,12-12 h晝夜條件下適應性飼養1周。將大鼠隨機分為5組(每組10只),分別為①假手術(Sham)組:使用異氟烷麻醉大鼠,但不阻塞其大腦中動脈;②I/R組:使用異氟烷麻醉大鼠,阻塞其大腦中動脈2 h致腦缺血后再恢復大腦中動脈血流灌注24 h,完成大鼠腦I/R損傷模型制備;③I/R+瑞馬唑侖(I/R+RE)組:經大鼠腹腔注射16 mg/kg瑞馬唑侖以后,立即制備大鼠腦I/R損傷模型;④I/R+RE+LY294002(LY)組:經大鼠側腦室注射50 mmol/L LY294002 10 μL,30 min后經腹腔注射16 mg/kg瑞馬唑侖,立即制備大鼠腦I/R損傷模型;⑤I/R+LY組:經大鼠側腦室注射50 mmol/L LY294002 10 μL,于30 min后制備大鼠腦I/R損傷模型。
1.3 各組大鼠血清IL-6、IL-1β和TNF-α水平分析
在大鼠腦I/R損傷模型制備完成以后,從各組大鼠當中分別取3只,每只于心臟左心室采血5~10 mL,將血液室溫靜置2 h以后離心取上清液,使用ELISA試劑盒測定各組大鼠血清中IL-6、IL-1β和TNF-α水平。
1.4 各組大鼠神經功能缺損評分及腦梗死體積比的計算
從每組大鼠中隨機選取3只大鼠,使用神經功能缺損評分量表[10]評估神經功能缺損情況。繼而使用異氟烷麻醉大鼠,并迅速取出其大腦置于直徑6 cm小皿中,-20 ℃冰箱冷凍30 min。再將大腦組織行2 mm厚冠狀切片,共切割5片,使用20 g/L的2,3,5-三苯基四氮唑氯化物溶液染色。染色組織拍照后使用Image J軟件分析缺血(白色)和正常(紅色)區域,然后計算腦梗死體積比。腦梗死體積比=腦梗死總體積/腦組織總體積×100%。
1.5 免疫印跡實驗檢測各組大鼠腦組織中AKT、p-AKT、NF-κB p65、p-NF-κB p65相對表達量
從每組大鼠中隨機選取3只大鼠,使用含磷酸酶抑制劑和蛋白酶抑制劑的組織裂解液裂解其梗死腦組織,提取腦組織梗死區域總蛋白,測定總蛋白濃度。然后上樣30 μg蛋白到100 g/L的聚丙烯酰胺凝膠上進行電泳分離,通過濕法轉移到PVDF膜上,并在室溫下使用50 g/L的脫脂奶粉密封PVDF膜1.5 h。之后向PVDF膜分別添加NF-κB p65、p-NF-κB p65、AKT一抗(1∶1 000)和p-AKT一抗(1∶2 000)、內參β-actin(1∶1 000),4 ℃下孵育過夜,再添加二抗室溫孵育1 h,滴加增強化學發光試劑,顯影儀曝光各蛋白條帶。以β-actin蛋白為內參照,并使用Image J軟件分析AKT、p-AKT、NF-κB p65、p-NF-κB p65相對表達量;同時計算p-AKT/AKT和p-NF-κB p65/NF-κB p65比值,分別表示AKT和NF-κB p65蛋白的活化程度。
1.6 免疫熒光法檢測各組大鼠腦組織細胞核內、外p-NF-κB p65表達
從每組大鼠中隨機選取3只大鼠,常規麻醉后處死,取整個腦組織以40 g/L多聚甲醛固定,切下紋狀體切面腦組織,并以石蠟包埋制成25 μm厚冠狀切片。切片以體積分數0.005曲拉通X-100滲透10 min,再用體積分數0.05的山羊血清封閉1 h,4 ℃下以稀釋的一抗p-NF-κB p65(1∶50)孵育過夜。隨后使用磷酸鹽溶液洗滌并添加二抗,室溫下避光孵育2 h,以4′,6-二脒基-2-苯基吲哚染色細胞核并封片,最后使用共聚焦激光顯微鏡觀察并拍照。
1.7 統計學方法
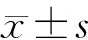
2 結 果
2.1 5組大鼠腦梗死體積比及神經功能缺損評分比較
5組大鼠的腦梗死體積比分別為0、(24.58±0.52)%、(6.22±0.70)%、(22.23±1.52)%和(26.28±0.96)%,各組間比較差異具有顯著性(F=537.30,P<0.05);5組大鼠的神經功能缺損評分則分別為0、(3.73±0.25)、(1.77±0.25)、(3.37±0.15)以及(4.10±0.10)分,各組間比較差異具有顯著性(F=271.30,P<0.05)。其中與Sham組相比,I/R組大鼠的腦梗死體積比及神經功能缺損評分顯著增高(P<0.05);與I/R組相比,I/R+RE組大鼠腦梗死體積比和神經功能缺損評分顯著下降(P<0.05);與I/R+RE組相比,I/R+RE+LY組和I/R+LY組大鼠腦梗死體積比和神經功能缺損評分均顯著增高(P<0.05)。
2.2 5組大鼠血清促炎細胞因子IL-6、IL-1β、TNF-α水平比較
ELISA檢測結果顯示,5組大鼠血清IL-6、IL-1β、TNF-α水平比較差異均有顯著性(F=72.41~209.30,P<0.05),其中與Sham組比較,I/R組大鼠血清IL-6、IL-1β、TNF-α的水平均顯著增高(P<0.05);與I/R組比較,I/R+RE組大鼠血清IL-6、IL-1β、TNF-α水平均顯著下降(P<0.05);與I/R+RE組比較,I/R+RE+LY組、I/R+LY組大鼠血清IL-6、IL-1β、TNF-α水平均顯著增高(P<0.05)。見表1。

表1 各組大鼠的血清中IL-6、IL-1β、TNF-α水平比較
2.3 5組大鼠腦組織AKT、p-AKT、NF-κB p65和p-NF-κB p65相對表達量比較
免疫印跡實驗的結果表明,各組大鼠腦組織中p-AKT、p-NF-κB p65相對表達量和p-AKT/AKT、p-NF-κB p65/NF-κB p65比值均有顯著差異(F=313.30~654.90,P<0.05),其中與Sham組相比較,I/R組大鼠p-AKT相對表達量和p-AKT/AKT比值顯著降低(P<0.05),p-NF-κB p65相對表達量以及p-NF-κB p65/NF-κB p65比值則顯著性增高(P<0.05)。與I/R組、I/R+RE+LY組和I/R+LY組相比,I/R+RE組大鼠p-AKT相對表達量和p-AKT/AKT比值均顯著增高(P<0.05),p-NF-κB p65相對表達量和p-NF-κB p65/NF-κB p65比值均顯著降低(P<0.05)。見圖1、表2。

A~E依次為Sham組、I/R組、I/R+RE組、I/R+RE+LY組、I/R+LY組

表2 各組大鼠腦組織AKT、p-AKT、NF-κB p65、p-NF-κB p65相對表達量及p-AKT/AKT、p-NF-κB p65/NF-κB p65比值比較
2.4 5組大鼠腦組織細胞核內、外p-NF-κB p65的表達比較
免疫熒光實驗結果顯示,Sham組大鼠p-NF-κB p65主要表達于細胞質,并且熒光強度較低。與Sham組相比,I/R組大鼠p-NF-κB p65從細胞質轉移到細胞核,熒光強度增高;與I/R組相比,I/R+RE組大鼠p-NF-κB p65從細胞核轉移到細胞質,熒光強度降低;與I/R+RE組相比,I/R+RE+LY組、I/R+LY組大鼠p-NF-κB p65均主要表達于細胞核,且熒光強度增高。見圖2。

A~E依次為Sham組、I/R組、I/R+RE組、I/R+RE+LY組、I/R+LY組,60倍
3 討 論
腦I/R損傷涉及機體一系列復雜的病理生理變化,如興奮毒性、氧化應激、能量代謝紊亂、炎癥反應和細胞凋亡等[11]。其中炎癥反應在腦I/R損傷過程中起重要作用[12]。因此,抑制炎癥反應是開發新的腦I/R損傷治療方法的關鍵[13]。瑞馬唑侖是一種新型苯二氮類短效鎮靜劑,已經被證明在腦I/R損傷中具有抗炎作用[9],但其確切的抗炎機制尚未完全明了。本研究使用腦I/R損傷大鼠模型研究瑞馬唑侖產生腦神經保護作用的機制,結果表明瑞馬唑侖通過減輕炎癥反應保護腦I/R損傷,其抗炎作用很大程度上是由AKT/NF-κB信號通路介導的。
本研究首先評估了瑞馬唑侖的腦神經保護作用。在實驗動物的腦I/R損傷模型中,腦梗死體積比和神經功能缺損評分是評估腦I/R損傷進展的主要參數,且神經功能缺損評分與腦梗死體積比相關[14]。因此,本研究通過測量腦梗死體積比和神經功能缺損評分評估瑞馬唑侖的體內療效,結果表明瑞馬唑侖能夠顯著降低腦梗死體積比和神經功能缺損評分,對腦I/R損傷具有保護作用。
其次,本研究觀察到瑞馬唑侖在腦I/R損傷中具有抗炎作用。炎癥細胞因子是腦I/R損傷后炎癥反應和神經元損傷的關鍵驅動因素[15]。在I/R損傷期間,大腦皮質免疫細胞激活,產生并釋放過量促炎細胞因子TNF-α、IL-1β和IL-6[16]。I/R中各種復雜因素導致血腦屏障被破壞,上述促炎細胞因子進入血液循環,血清和腦脊液中TNF-α和IL-1β水平升高,使得小膠質細胞活化,促進巨噬細胞和中性粒細胞聚集并向大腦缺血區域浸潤,從而進一步破壞腦組織和加速神經元丟失[17]。據報道,血清及腦脊液中IL-6水平升高與I/R損傷所致的神經系統惡化相關[18-19]。本研究使用ELISA試劑盒,測定各組大鼠血清促炎細胞因子TNF-α、IL-1β和IL-6水平,結果顯示與I/R組相比,I/R+RE組促炎細胞因子水平顯著降低,表明瑞馬唑侖通過減輕腦I/R時炎癥反應發揮腦神經保護作用。
最后,本研究探討了瑞馬唑侖在腦I/R損傷中的抗炎機制。NF-κB是炎癥中重要的二聚轉錄因子家族,p65-p50異二聚體是NF-κB最常見的種類,而具有多功能結構域的p65是NF-κB最重要的亞基[20]。腦I/R期間,缺氧、過量活性氧和炎癥介質誘導p65磷酸化,使其從細胞質易位進入細胞核,并特異性結合炎性細胞因子的啟動子,從而誘導IL-1β、IL-6和TNF-α的表達,導致神經元死亡[21]。既往研究表明,PI3K/AKT的激活對腦I/R具有腦神經保護作用,且PI3K/AKT激活可抑制NF-κB通路活化[22]。根據報道,咪達唑侖(化合物結構與瑞馬唑侖類似)能夠上調神經元細胞中P-AKT的表達[23],瑞馬唑侖能夠下調神經性疼痛和膠質瘤中NF-κB的表達[8,24]。本研究通過免疫印跡檢測了各組大鼠腦組織中AKT、p-AKT、NF-κB p65和p-NF-κB p65的表達水平,并使用免疫熒光法檢測了各組大鼠腦組織細胞核內、外p-NF-κB p65的表達,結果表明,腦I/R損傷以后,瑞馬唑侖上調了腦組織中p-AKT表達水平,下調了p-NF-κB p65表達水平,并使得p-NF-κB p65從細胞核轉移到細胞質,減少了其細胞核內的表達。為了闡明AKT/NF-κB信號通路是否參與瑞馬唑侖在腦I/R損傷中的抗炎作用,本研究使用LY294002抑制PI3K/AKT的功能,結果顯示,LY294002能夠下調腦組織中p-AKT表達水平,上調p-NF-κB p65表達水平,增加了p-NF-κB p65的細核內表達,并逆轉了瑞馬唑侖的腦神經保護作用和抗炎作用。這些結果表明,AKT/NF-κB信號通路在瑞馬唑侖對腦I/R損傷的腦神經保護和抗炎作用中起著重要作用。
綜上所述,瑞馬唑侖對腦I/R損傷具有保護作用,其保護機制可能與AKT/NF-κB信號通路介導的抗炎作用相關。
倫理批準和動物權利聲明:本研究涉及的所有動物實驗均已通過青島大學醫學部倫理委員會的審核批準(文件號AHQU-MAL2021-1105)。所有實驗過程均遵照美國國立衛生院出版的《實驗動物合理和使用指南》的條例進行。
作者聲明:段梅、劉佳、王士雷參與了研究設計;段梅、劉佳、張靜、孫曉麗、王士雷參與了論文的寫作和修改。所有作者均閱讀并同意發表該論文,且均聲明不存在利益沖突。
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
昆明醫科大學學報(2020年12期)2021-01-26 00:44:04
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
豬業科學(2018年8期)2018-09-28 01:27:38
中成藥(2017年8期)2017-11-22 03:18:47
川北醫學院學報(2015年5期)2015-12-05 08:22:29
