對GB/T 8313—2018標準中茶多酚重復性規定的探討
2023-10-21 07:04:22王小樂
中國茶葉 2023年10期
關鍵詞:標準
王小樂
漢中市質量技術監督檢驗檢測中心,陜西 漢中 723000
茶多酚是茶葉在生長過程中自然形成的天然物質,是決定茶葉滋味品質的關鍵成分。同時茶多酚具有抗氧化、抗輻射等保健功效,近年來倍受商家及消費者關注。我國頒布實施的茶葉及茶制品國家標準均對產品中茶多酚含量及檢測方法有明確規定。我國推薦的現行有效測定茶多酚含量的方法是《茶葉中茶多酚和兒茶素類含量的檢測方法》(GB/T 8313—2018,以下簡稱“方法”)中的第二法。該方法為茶葉及其制品在產品開發、生產加工及市場銷售階段提供茶多酚含量測定的技術支持。該方法中檢測茶多酚含量的重復性規定為“同一樣品茶多酚含量的兩次測定值(化學分析中稱作“平行樣”)相對誤差應≤5%”[1]。截至目前,未有相關文獻對檢測茶多酚含量的重復性規定值進行分析研究。本文從重復性定義和數學模型入手,找出影響茶多酚測定結果的因素來源,并通過不確定度量化評定,對其重復性規定的合理性進行驗證。
1 重復性的測量條件
在化學分析試驗中,重復性體現測試結果的一致程度。測量重復性,指在一組相同測量程序、相同操作者、相同測量系統、相同操作條件和相同地點,并在短時間內對同一或相類似被測對象重復測量的測量精密度[2]。如果重復性不能滿足要求,說明測得結果不滿足一致程度。
1.1 相同測量程序
方法中對原理、儀器、試劑、操作方法、測定及結果計算作出明確規定。平行樣操作的所有程序應是不變的,可視為相同。
1.2 相同操作者
平行樣由一人獨立操作完成,整個測試及結果計算過程,可視為相同。
1.3 相同測量系統
檢測中所用儀器包括天平、水浴鍋、離心機、分光光度計,在試驗過程中均由同一設備完成。但由于平行樣稱量、提取、稀釋、測定等操作是獨立進行的,儀器的示值誤差和重復性誤差會帶來測定結果的不一致。過程中還需要使用玻璃量器,如:移液管、容量瓶。這些器具引入的體積、溫度和讀數誤差也應一并考慮,所以測量系統不能視為完全相同。
1.4 相同操作條件
方法中規定了水浴溫度和時間、離心機轉數和時間、加入福林酚試劑的反應時間以及測定前靜置時間。從理論上講,整個試驗過程是在相同操作條件下進行的,但實際操作過程中由于該試驗操作步驟繁瑣,人工操作很難保證每個步驟所用時間完全相同,平行樣在操作過程中或多或少會引入時間和溫度上的波動,所以操作條件不能視為完全相同。
1.5 相同地點
在相同的實驗室內進行測試,可視為相同。
1.6 在短時間內重復測量
方法中多處對時間有嚴格要求,整個過程滿足在短時間內重復測試,可視為相同。
通過對重復性測量條件的分析,可以看出測量系統和操作條件是引入誤差的主要來源。
2 評估模型及建模分析
2.1 評估數學模型
式中:x1、x2為單次測定茶多酚含量(%);x為茶多酚含量的平均值;c1為測試液中茶多酚的質量濃度(μg/mL);v為提取液定容體積(mL);m1為茶樣質量(g);m0為茶樣干物質百分含量(%);d為稀釋因子。
2.2 數學模型分析
從式(1)中可知,有x1、x2、這3 個變量,x1和x2為平行測定的兩份樣品,分別由試驗測定并通過式(2)計算得出,由(x1+x2)/2 計算得出。理想情況下x1=x2=,但試驗中不可避免隨機誤差和系統誤差的存在。
假設對同一茶樣同時測定n 份,(x1,xn)為茶多酚含量測定結果區間,為中位值,這樣就形成了一個正態分布,(x1,xn)為的分散程度。要滿足相對誤差≤5%,(x1,xn)的分散程度越小越好。
由(x1+…+xn)/n 計算得到,其值大小主要由茶葉特質決定。因原料鮮葉和生產工藝的不同,茶葉中茶多酚含量差異從3%到34%不等[3]。
從式(2)中可以看出,茶多酚的含量與c1、m1、m0、v、d這5 個參數相關,v和d是固定值,分別代表提取定容體積、稀釋倍數,但在實際操作中體積和溫度會引入隨機誤差,所以式(2)中5個參數都會對測試結果茶多酚含量產生影響,這也是造成x1≠x2、x1≠xn的原因所在。
通過圖1清晰地反映出各參數中影響茶多酚測定結果的來源,通常量化采用不確定度進行評定[4-7]。
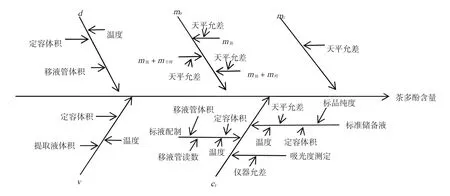
圖1 測定過程中各參數對測定結果影響的魚骨圖
3 試驗方法
3.1 原理
以甲醇水溶液作為提取液,對磨碎茶葉樣品進行浸提,待測液在堿性條件下,茶多酚中-OH基團被福林酚氧化顯藍色,在765 nm 波長下,以沒食子酸作標準曲線,測定茶多酚含量。
3.2 操作方法
3.2.1 樣品母液制備
精確稱取0.2 g 茶樣于離心管中,加入70%甲醇水溶液5 mL 后,水浴加熱(70 ℃)10 min,邊加熱邊攪拌。待冷卻后以3 500 r/min 離心10 min,移出上清液。殘渣按上述操作再重復提取1次,合并上清液定容至10 mL。
3.2.2 標準儲備液配制
精確稱取0.110 g 沒食子酸標準品,加70%甲醇水溶液溶解,定容于100 mL容量瓶。
3.2.3 沒食子酸工作液配制
分別移取1.0、2.0、3.0、4.0、5.0 mL 沒食子酸標準儲備液,加水定容于100 mL容量瓶。
3.2.4 樣品測試液配制
移取1.0 mL母液加水定容于100 mL容量瓶。
3.2.5 測定
移取沒食子酸工作液、水及樣品測試液各1.0 mL,分別加入5.0 mL 福林酚試劑,搖勻放置5 min,加入4.0 mL7.5%碳酸鈉溶液搖勻,放置1 h,用1 cm比色皿測定吸光度。
4 不確定度來源及評定
4.1 不確定度來源
通過對數學模型的分析,梳理出測定茶多酚含量的不確定度分量、不確定度具體來源及影響因素(表1)。
用戶如果忘記密碼,可以使用注冊的手機號找回密碼。具體實現方式是通過給注冊手機號發送驗證碼,輸入驗證碼,再輸入新密碼的方式來找回密碼。
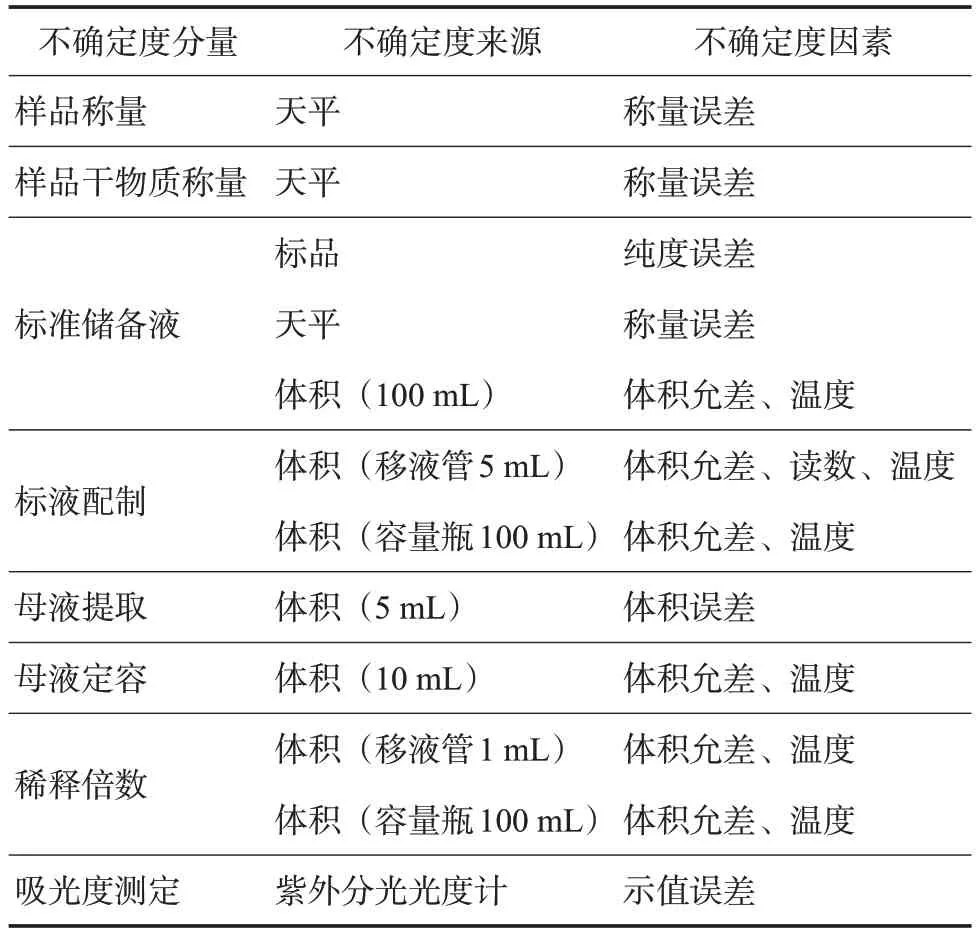
表1 不確定度來源分析
4.2 不確定度分量評定
4.2.1 樣品稱量引入的不確定度ur(m樣)
稱取0.2 g 茶樣,應選I級天平稱量,0~50 g稱量范圍允許誤差為±0.000 5 g[8],按照均勻分布,樣品稱量的相對標準不確定度為。
4.2.2 樣品干物質引入的不確定度ur(m干)
干物質含量測定采用恒重法,采用天平稱重[9]。稱取5.0 g 茶樣,參照4.2.1 中天平允差數據及分布,樣品干物質的相對不確定度為。
4.2.3 標品純度引入的不確定度ur(p)
沒食子酸標品的純度為99.47%,擴展不確定度為0.30%,按照均勻分布,沒食子酸純度的相對不確定度為
4.2.4 標品稱量引入的不確定度ur(m標)
稱取0.110 g沒食子酸標品,參照4.2.1中天平允差數據及分布,標品稱量的相對標準不確定度為。
(1)本試驗全部選用A級玻璃量器,標品定容于100 mL 容量瓶,體積允差為±0.10 mL[10],按照三角分布,體積校準的相對標準不確定度為。
(2)方法中未對標品定容試劑做具體規定,考慮標品制備和樣品制備保持一致原則,選擇用70%的甲醇水溶液定容。玻璃量器在20 ℃校準,而實驗室溫度在(20±4)℃范圍波動。水的膨脹系數為2.08×10-4/℃,甲醇的膨脹系數為1.18×10-3/℃,70%甲醇水溶液的膨脹系數為0.7×1.18×10-3+0.3×2.08×10-4= 8.88×10-4/℃。依據膨脹效應體積變化為±(100×4×8.88×10-4)mL=±0.355 2 mL,按照均勻分布,溶液溫度引入的體積變化相對標準不確定度為。
標品定容的合成相對標準不確定度見式(3)。
4.2.6 標液配制引入的不確定度ur(v標配)
(1)標液配制使用5 mL刻度移液管和100 mL容量瓶,體積允差分別為±0.025 mL、±0.10 mL,參照4.2.5(1)中方法計算,移液管體積校準的相對標準不確定度為2.04×10-3,容量瓶體積校準的相對標準不確定度為4.08×10-4。
(2)移取液體會因溫度變化帶來溶劑體積變化,參照4.2.5(2)中70%甲醇水溶液的膨脹系數8.88×10-4/℃,移液管體積產生的變化為±(5×4×8.88×10-4)mL=±1.78×10-2mL,按照均勻分布,移取溶液時溫度引入的體積變化相對標準不確定度為2.06×10-3。定容所用試劑為純水,水的膨脹系數為2.08×10-4/℃,體積產生的變化為±(100×4×2.08×10-4)mL=±8.32×10-2mL,定容時溶液溫度引入的體積變化相對標準不確定度為4.8×10-4。
(3)配制標準系列時,移液管移取沒食子酸標準儲備液會引入讀數誤差,一般估計為±0.01 mL,按照三角分布,讀數的相對不確定度為。
標液配制的合成相對標準不確定度見式(4)。
4.2.7 母液提取引入的不確定度ur(v提)
母液提取離心后,由人工手動轉移上清液,該步驟不能保證每次移取體積相同,根據經驗引入的不確定度估計為0.05 mL,故a = 0.025 mL,按照三角分布,提取液移取的相對標準不確定度為。
4.2.8 母液定容引入的不確定度ur(v提定)
(1)提取液定容于10 mL 容量瓶,體積允差為±0.020 mL,按照三角分布,體積校準的相對標準不確定度為8.16×10-4。
(2)母液用70%甲醇水溶液定容,體積產生的變化為±0.035 5 mL,按照均勻分布,溶液溫度引入的體積變化相對標準不確定度為2.05×10-3。母液定容的合成相對標準不確定度見式(5)。
4.2.9 稀釋倍數引入的不確定度ur(v稀)
(1)配制樣品測試液使用1 mL移液管和100 mL容量瓶,體積允差分別為±0.008 mL、±0.10 mL,按照三角分布,移液管體積校準的相對標準不確定度為3.27×10-3,容量瓶體積校準的相對標準不確定度為4.08×10-4。
(2) 移液管因溫度變化產生的體積變化為±(1×4×2.08×10-4)mL=±8.32×10-4mL,按照均勻分布,移液管溫度變化的相對標準不確定度為4.8×10-4,參照4.2.6(2)中容量瓶溫度引入的體積變化相對標準不確定度為4.8×10-4。
稀釋倍數的合成相對標準不確定度見式(6)。
4.2.10 紫外分光光度計引入的不確定度ur(A)
該試驗采用島津UV-2600紫外分光光度計,等級為Ⅱ級,波長765 nm屬于B段,該段的透射比最大允許誤差±0.005[11],按照均勻分布,紫外分光光度計的標準不確定度為。
4.3 合成不確定度及擴展不確定度
將4.2所有不確定度分量按質量、體積、吸光度、標品純度分別擬合,最終計算合成標準不確定度:其中。按95%置信概率,取包含因子k=2,擴展不確定度U=k×uc=0.015。各項不確定度的貢獻率見表2。
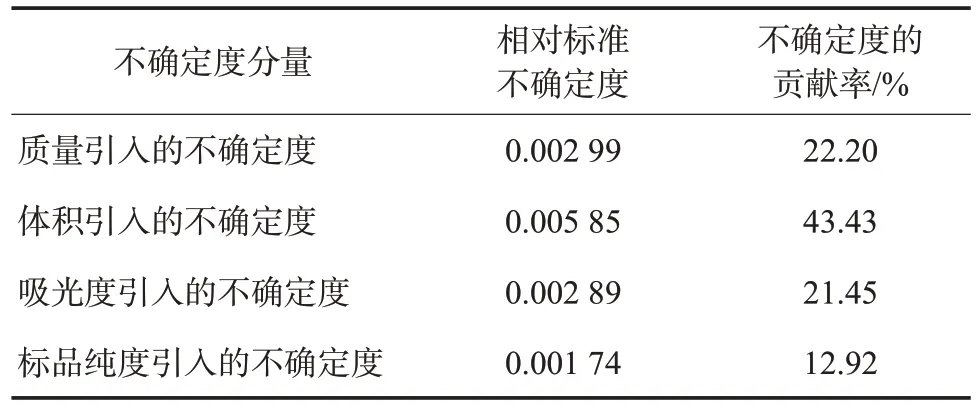
表2 茶多酚測定的相對不確定度分量
5 分析討論
從表2不確定度的貢獻率可以看出,體積引入的不確定度最大。這與試驗過程復雜,使用大量玻璃量器有直接關系。該試驗選用I級電子天平和Ⅱ級紫外分光光度計,儀器精度等級較高,但質量和吸光度的不確定度貢獻率依然高達22.20%、21.45%。標品純度引入的不確定度最小,按照小于最大分量三分之一可以不予考慮[12]。
uc值為測量茶多酚含量引入的合成標準不確定度,具有分散性和非負數特征。假設被測量茶多酚含量的可能值在[y-uc,y+uc]區間內,為使uc與y量綱相同,uc值乘以100%為0.74%。分別以y=20%,y=30%代入式(1)計算。
當茶葉中茶多酚含量y=20%,
當茶葉中茶多酚含量y=30%,
通過代入不同的茶多酚含量值計算平行樣相對誤差。茶多酚含量為20%的茶樣,相對誤差>5%,不滿足重復性結果一致程度;茶多酚含量為30%的茶樣,相對誤差<5%,滿足重復性結果一致程度。試驗證明,在平行樣分散度相同的情況下,茶多酚含量值越大,相對誤差越小。
6 結論
本文通過對《茶葉中茶多酚和兒茶素類含量的檢測方法》(GB/T 8313—2018)中茶多酚含量測定的全面分析得出,測試結果的重復性與平行樣分散度、茶葉自身的茶多酚含量相關。要得到較小的分散度,盡可能選用玻璃量器A級中檢定或校準容量偏差較小的產品。選用靈敏度較高的儀器,并定期做好電子天平和紫外分光光度計的檢定校準及日常維護。操作人員應熟悉操作方法,在試驗過程中保持操作順暢,同一過程盡可能所用時間相同。同時,能否獲得滿意的重復性結果與茶葉中茶多酚含量關系密切。由于茶樹品種和生長環境的不同,以及六大茶類不同的生產工藝,使得茶葉中茶多酚含量差異較大,必須綜合考慮茶葉自身的特點,針對不同茶多酚含量科學制定相對誤差值,才能對其測試結果一致性進行有效判定。
2023年6月中國茶葉出口各國和地區銷量統計 t
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
