馬拉硫磷的納米金適配體比色傳感檢測方法
2023-10-21 03:14:38田恒旗楊海濤李兆周吳佳蓓劉浩宇常云鶴陳秀金
食品科學 2023年18期
田恒旗,王 耀,,*,楊海濤,李兆周,吳佳蓓,王 琳,劉浩宇,常云鶴,陳秀金,*
(1.河南科技大學食品與生物工程學院,河南 洛陽 471023;2.河南宜測科技有限公司,河南 鄭州 451162;3.貴陽學院食品與制藥工程學院,貴州 貴陽 550005)
農藥在保障農產品產量和質量的同時,其本身也是一把雙刃劍,隨著農藥的種類、使用量以及農藥適用范圍的日益增加,相關食品安全事件頻頻發生,農藥殘留成為了影響農產品安全問題的一個重要因素[1-2]。馬拉硫磷是果蔬種植過程中常用的殺蟲劑,也常作為谷物儲藏的保護劑。馬拉硫磷雖屬于低毒性化合物[3],但長期不合理的使用會導致環境及食品中馬拉硫磷殘留量超標,經食物鏈的富集進入人體內,通過抑制乙酰膽堿酯酶的活性,造成肝臟以及中樞神經系統受損,嚴重的可導致中毒死亡[4]。長時間暴露于含馬拉硫磷的環境中也可能引發心肌炎、肺水腫、胰腺炎等并發癥[5]。
傳統的馬拉硫磷農藥殘留檢測方法多為儀器檢測法[6],如利用質譜儀或色譜儀檢測農藥殘留[7],其準確性高、穩定性好,可同時檢測多種成分[8]。但是傳統檢測方法因儀器價格昂貴、樣品處理繁瑣、經濟成本高,多作為確證手段使用,無法滿足現場即時檢測需要[9]。目前馬拉硫磷的快速檢測方法主要有酶抑制法和免疫分析法[10],酶抑制法的檢測成本較低,易于操作,但是酶不穩定、容易失活,導致誤差很大[11]。免疫分析法的特異性強,但是特異性抗體制備周期長、成本較高[12]。
適配體是通過體外篩選技術-指數富集配體系統進化技術(systematic evolution of ligands by exponential enrichment,SELEX),從隨機單鏈寡聚核苷酸文庫中得到的一種能特異結合待測靶標(如蛋白質或其他的小分子物質)的單鏈核苷酸[13]。與傳統的抗體相比,適配體具有易合成、可修飾性強、結構簡單、性質穩定等優勢[14-15]。目前,已有多種農藥被篩選出高親和力和強特異性的適配體[16],在此基礎上,也建立了包括比色法[17]、熒光法[18]、電化學法[19]在內的多種農藥殘留適配體傳感檢測方法,推動了農藥殘留快速檢測技術的發展[20]。
本研究以馬拉硫磷特異性適配體作為識別元件、基于納米金(gold nanoparticles,AuNPs)作為顏色指示劑,建立了馬拉硫磷的適配體比色傳感檢測方法。本方法原理如圖1所示,AuNPs是穩定的膠體溶液[21],在高鹽環境下AuNPs會發生聚集,顏色和紫外吸收光譜會發生變化,若在體系中加入適配體,通過靜電吸附作用,適配體會結合到AuNPs表面,保護AuNPs在溶液中保持分散狀態,不發生聚集[22],體系顏色也不發生變化,當加入馬拉硫磷后,適配體與馬拉硫磷的高親和力使得適配體會優先與馬拉硫磷結合,AuNPs失去了適配體的保護,在高鹽濃度下會發生聚集,溶液的顏色由紅色逐漸變為藍紫色,顏色的變化程度與添加的馬拉硫磷質量濃度呈正相關。
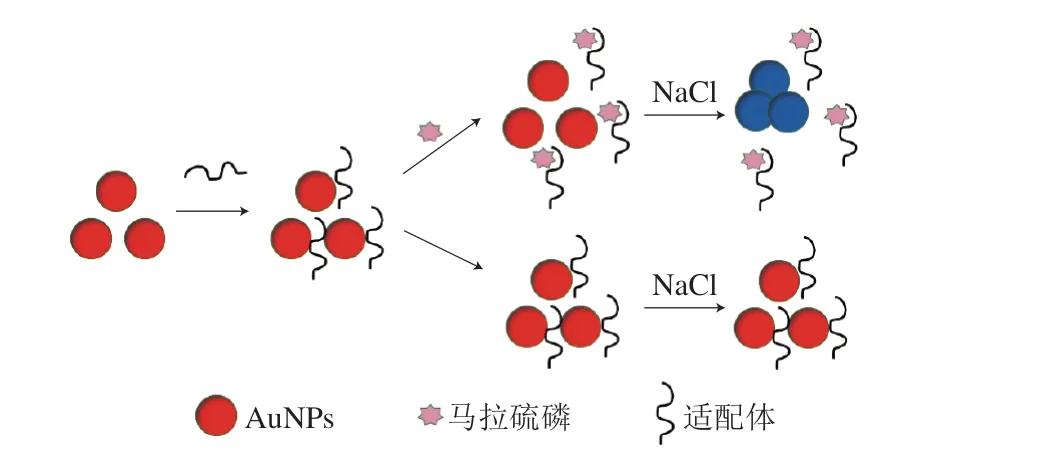
圖1 AuNPs-適配體比色傳感法原理圖Fig.1 Schematic diagram of the principle of colorimetric sensing method using AuNPs-based aptamer
1 材料與方法
1.1 材料與試劑
生菜和蘋果購于本地超市。
氯金酸(HAuCl4)、檸檬酸鈉、EDTA、K2CO3、馬拉硫磷、毒死蜱、樂果、丙溴磷、倍硫磷、吡蟲啉、敵敵畏、對硫磷標準品 上海阿拉丁生化科技股份有限公司;甲醇、乙腈、NaCl、HCl、單寧酸(均為分析純)天津市德恩化學試劑有限公司。實驗用水均為超純水(18.2 MΩ·cm)。
馬拉硫磷適配體序列參照Williams等[23]的研究,并由生工生物工程(上海)股份有限公司合成,序列如下:5’-TGT ACC GTC TGA GCG ATT CGT ACT ATG GTA TCC GAG AGG CCTACG GAA TTG TTG TAC AGC CAG TCA GTG TTA AGG AGT GC-3’。
1.2 儀器與設備
透射電子顯微鏡 日立科學儀器有限公司;UV-2600紫外-可見分光光度計 日本島津公司;氣相色譜-串聯三重四極桿質譜聯用儀 賽默飛世爾科技有限公司;H1650R臺式高速冷凍離心機 湖南湘儀實驗室儀器開發有限公司;pHS-3C pH計 上海雷磁儀器廠;Vortex 1渦旋混均器 德國IKA公司;JJ224BF型電子分析天平 賽多利斯科學儀器有限公司。
1.3 方法
1.3.1 AuNPs的制備
使用單寧酸和檸檬酸為還原劑合成AuNPs[24]。首先,在攪拌下將1 mL質量分數為1%的HAuCl4和79 mL超純水加入到一個燒瓶中,然后在攪拌下將4 mL質量分數為1%的檸檬酸鈉、0.1 mL質量分數為1%的單寧酸、0.1 mL濃度為25 mmol/L的K2CO3和15.8 mL超純水加入到另一個燒瓶中,將上述兩種制備的溶液分別加熱至60 ℃并保持30 min,然后在高速攪拌下混合在一起,直至顏色變為酒紅色且不再變化,然后冷卻至室溫,將溶液儲存在4 ℃冰箱中。
1.3.2 AuNPs-適配體比色法檢測馬拉硫磷
用1×TE緩沖液(10 mmol/L Tris-HCl、1 mmol/L EDTA,pH 8.0)配制1 μmol/L馬拉硫磷適配體溶液,然后將30 μL適配體溶液和150 μL AuNPs充分混勻,孵育10 min,加入30 μL馬拉硫磷標準品溶液混勻,孵育10 min,再加入30 μL濃度為500 mmol/L的NaCl溶液充分混勻,反應10 min,裸眼觀察溶液的顏色變化,并用UV-2600紫外-可見分光光度計掃描350~750 nm的紫外吸收光譜,記錄峰值變化,配制不同質量濃度馬拉硫磷標準溶液(50、100、200、300、400、800、1 500 ng/mL),用于繪制標準曲線,所有測定均在室溫下進行。
1.3.3 特異性鑒定
配制800 ng/mL的馬拉硫磷、樂果、乙酰甲胺磷、毒死蜱、敵敵畏、草甘膦、吡蟲啉、倍硫磷溶液,按照AuNPs-適配體比色傳感法檢測馬拉硫磷的步驟,分別進行適配體的特異性鑒定,并取等體積的緩沖液作為陰性對照。
1.3.4 實際樣品檢測
粉碎并稱取5.0 g具有代表性的果蔬樣本并加工成均質的勻漿,加入20 mL濃度為10 mmol/L的Tris-HCl緩沖溶液與其混合,然后置于振蕩器上劇烈振蕩10 min,之后室溫下8 000 r/min離心20 min,以除去固體物質。然后使用0.22 μm的微孔濾膜過濾上清液,將過濾后的上清液作為提取液。將馬拉硫磷標準品添加到提取液中,制成不同質量濃度(200、400、800 ng/mL)的加標檢測液。使用本方法和國標法進行實際樣品中馬拉硫磷的檢測并將檢測結果進行對比。
1.3.5 儀器驗證
參照國家標準方法(GB 23200.113—2018《植物源性食品中208 種農藥及其代謝物殘留量的測定 氣相色譜-質譜聯用法》)[25],利用氣相色譜-質譜聯用法對加標樣品進行檢測,條件如下:色譜柱溫度40 ℃保持1 min,然后以40 ℃/min程序升溫至120 ℃,再以5 ℃/min升溫至240 ℃,再以12 ℃/min升溫至300 ℃,保持6 min,進樣量為1 μL,流速為1.0 mL/min。
1.4 數據統計與圖表繪制
采用Origin 2021軟件進行數據統計及圖表繪制,每組實驗重復3 次平行實驗,取平均值作為實驗數據。
2 結果與分析
2.1 AuNPs的表征
制備的AuNPs為顏色鮮艷的酒紅色膠體溶液,紫外表征發現其特征吸收峰在518 nm波長處,峰形較為尖銳(圖2),這表明制備的AuNPs粒徑均一,具有較好的性能。用透射電鏡對制備的AuNPs的外觀形貌進行表征(圖3),可以看出AuNPs呈現統一的圓球形,粒徑較為均一,約為10 nm。正常情況下為分散狀態(圖3A),而在加入NaCl溶液后會發生聚集(圖3B)。綜上所述,本研究合成的AuNPs在水中呈現膠體狀,具有均有的大小和形狀,分散性良好,性質穩定[26]。
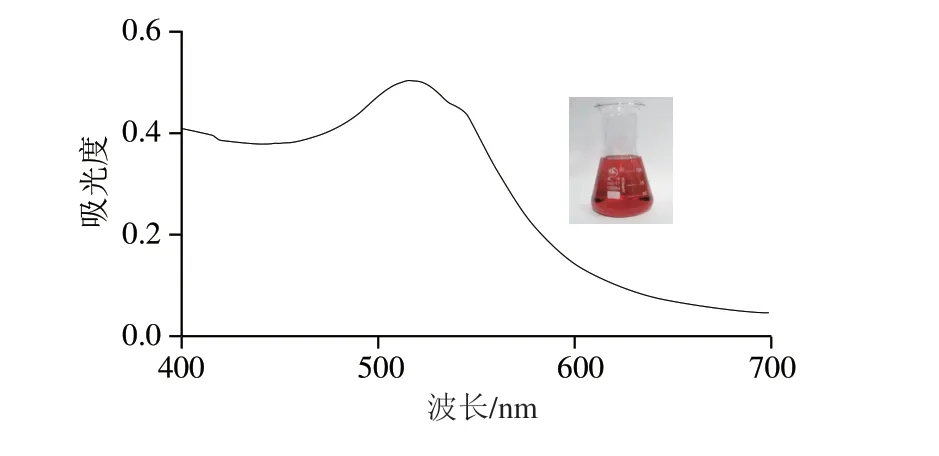
圖2 AuNPs的紫外表征Fig.2 Ultraviolet absorption spectrum of AuNPs

圖3 AuNPs的透射電鏡表征Fig.3 Transmission electron microscopic image of AuNPs
2.2 檢測體系優化
2.2.1 AuNPs體積的優化
AuNPs是整個檢測體系的顏色指示劑,其體積在整個反應體系中的占比直接影響方法的靈敏度,為獲得裸眼觀察的最佳指示顏色深度,通過向溶液中加入30 μL的馬拉硫磷適配體溶液、馬拉硫磷標準品溶液和500 mmol/L N a C l 溶液,優化A u N P s 在整個反應體積中的占比(40.0%、50.0%、62.5%、70.0%),結果見圖4,當AuNPs體積分數為40%、50%和62.5%時,體系可以由紅色變藍色,但是體積分數為40%和50%時,顏色變化不明顯,體積分數為62.5%時,顏色變化較為明顯。因此選擇AuNPs的體積分數為62.5%,即加入150 μL AuNPs溶液。
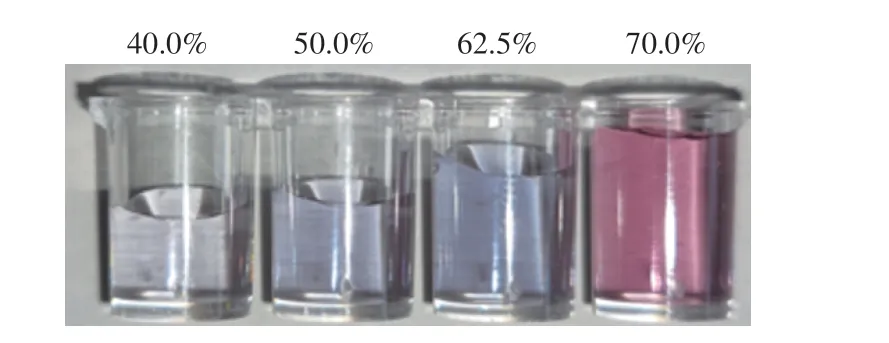
圖4 AuNPs的體積優化Fig.4 Optimization of volume of AuNPs used
2.2.2 NaCl濃度的優化
AuNPs會在高鹽環境下發生聚集,隨著NaCl濃度的增加,AuNPs的聚集程度也隨之增加,AuNPs的顏色發生明顯變化,由酒紅色逐漸變為藍紫色。當NaCl濃度過大時,在沒有靶標的情況下AuNPs也可能發生聚集;當NaCl濃度過小時,AuNPs可能只發生部分聚集從而影響試驗的結果,因此本研究對NaCl濃度進行優化[27]。在AuNPs的最佳反應體積下(150 μL),用超純水代替適配體和靶標,然后加入30 μL不同濃度(0、50、200、350、500、650、800、950 mmol/L)的NaCl溶液,充分混勻,靜置10 min后觀察溶液的顏色并使用紫外分光光度計進行吸光度的測定。
如圖5所示,當NaCl濃度低于500 mmol/L時,AuNPs呈紅色,表明此時的NaCl濃度較低,無法使AuNPs發生聚集;當NaCl濃度為500 mmol/L時,AuNPs發生聚集,顏色由紅色變為藍色。之后隨著NaCl濃度的繼續增加,AuNPs的顏色不再發生明顯變化。但是單以顏色變化不能夠準確判定出AuNPs聚集完成情況,需要根據紫外吸收光譜的變化進行判定。由圖5可以看出,隨著NaCl濃度增加,AuNPs在518 nm波長處的吸收峰值逐漸變小,并且在620 nm波長處出現了一個新的特征吸收峰,這是因為AuNPs發生聚集導致等離子發生了共振變化,使得AuNPs的特征吸收峰向右發生了偏移。因此,需要根據A620nm/A518nm值判定AuNPs的聚集程度,結果如圖6所示,當NaCl濃度為500 mmol/L時,A620nm/A518nm值突然增大,之后隨著NaCl濃度的繼續增加,不再發生明顯變化,因此NaCl的最佳濃度為500 mmol/L。
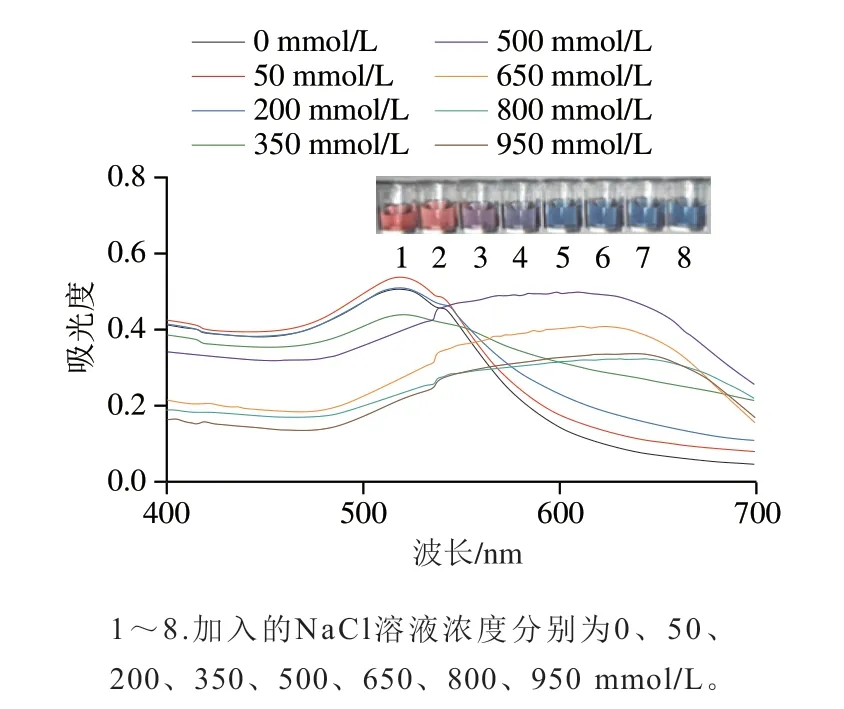
圖5 NaCl的濃度優化Fig.5 Optimization of NaCl concentration
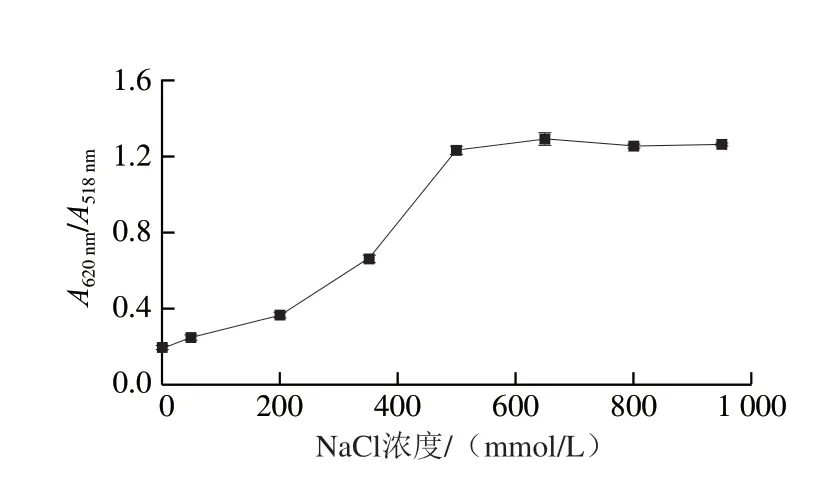
圖6 不同NaCl濃度下A620 nm/A518 nm值Fig.6 A620 nm/A518 nm ratio at different NaCl concentrations
2.2.3 適配體濃度的優化
當AuNPs溶液中不存在適配體時,AuNPs在高鹽環境下會發生聚集,AuNPs由紅色變為藍紫色;當加入的適配體濃度較低時,通過靜電吸附作用,低濃度的適配體可以吸附在AuNPs表面,但因為濃度較低,不能夠將AuNPs的表面完全包裹,仍然有一部分AuNPs呈單一分散狀態,此時AuNPs溶液會由藍色變為藍紫色;當適配體濃度為1 μmol/L時,適配體可以保護AuNPs不發生聚集現象,此時AuNPs溶液呈現酒紅色,之后隨著適配體濃度的繼續增加,不再發生明顯變化。如圖7所示,當適配體濃度為1 μmol/L時,A620nm/A518nm值也不再隨著適配體濃度的增加而改變,說明在500 mmol/L的NaCl濃度下,1 μmol/L的適配體已經將AuNPs的表面完全包裹,使其結構更加穩定,不再受高NaCl濃度的影響發生聚集現象。
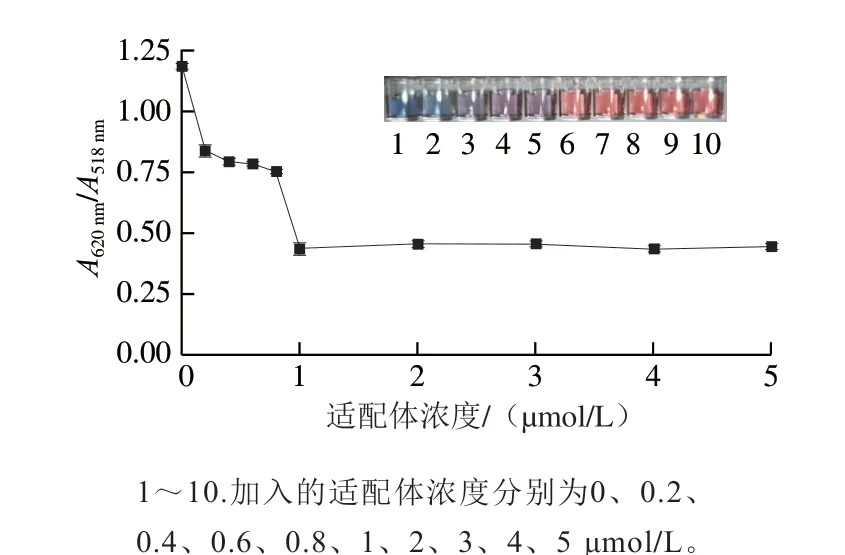
圖7 不同適配體濃度下A620 nm/A518 nm值Fig.7 A620 nm/A518 nm ratio at different aptamer concentrations
2.2.4 反應時間的優化
AuNPs在高濃度NaCl溶液中的聚集需要一定時間,在達到穩定狀態之前,顏色會不斷發生變化,與之相應的A518nm也會不斷發生變化。為了使檢測更為準確,減小誤差,需要研究體系顏色從不斷發生變化到達到穩定狀態所需要的時間。如圖8所示,當NaCl濃度為500 mmol/L時,A518nm在10 min之前,隨著反應時間的延長吸光度不斷降低;而在10 min后,隨著反應時間延長,吸光度不再發生明顯變化。因此,AuNPs與500 mmol/L的NaCl溶液的最佳反應時間為10 min。
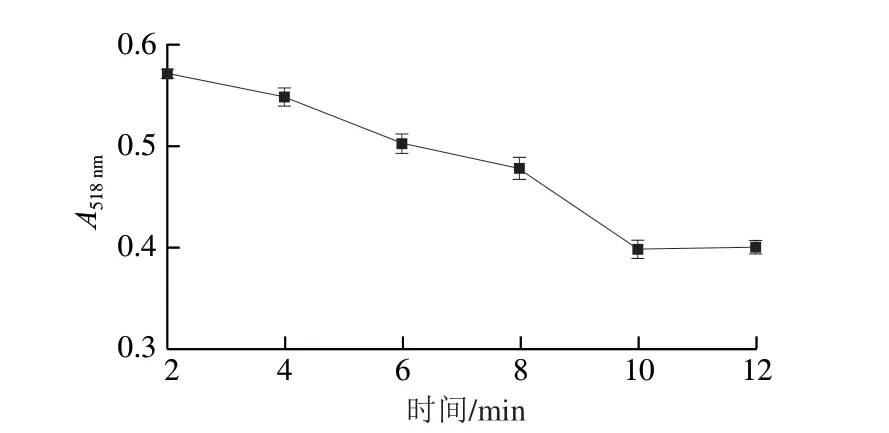
圖8 AuNPs與NaCl不同反應時間下A518 nm值Fig.8 Change in A518 nm as a function of reaction time between AuNPs and NaCl
如圖9所示,隨著AuNPs與適配體孵育時間延長,A518nm也隨之增加,在10 min時吸光度達到最大。然后隨著時間延長,吸光度開始出現一段下降趨勢。這表明時間較短時,適配體還沒有完全吸附在AuNPs的表面,存在一部分分散的AuNPs;適配體作為單鏈DNA序列,反應時間過長時,序列的質量在室溫下會有所下降,可能會導致部分AuNPs不再處于聚集狀態。綜上所述,AuNPs與適配體的最優反應時間為10 min。
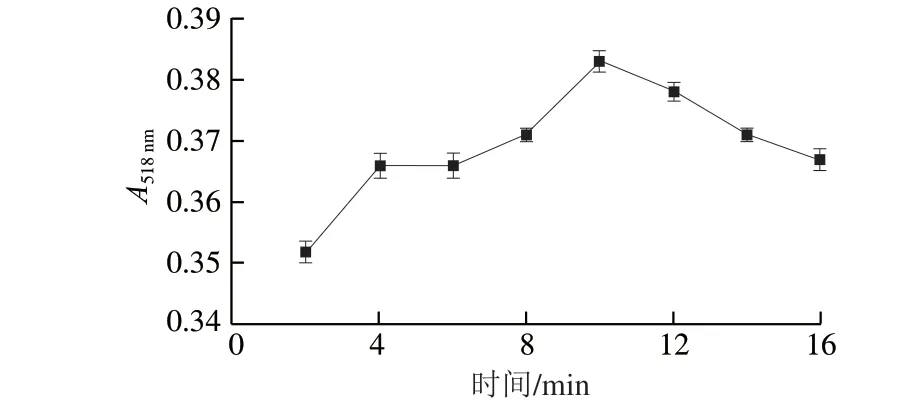
圖9 AuNPs與適配體不同反應時間下A518 nm值Fig.9 Change in A518 nm as a function of reaction time between AuNPs and aptamer
2.3 靈敏度檢測
在最佳的條件下,建立了AuNPs-適配體比色傳感法定量檢測馬拉硫磷的方法,為減弱基質效應影響,采用與實際樣品相同的加標條件繪制了基質標準曲線。如圖10所示,隨著馬拉硫磷質量濃度升高,溶液顏色逐漸由紅變藍,在馬拉硫磷質量濃度范圍為50~1 500 ng/mL時,A620nm/A518nm值與馬拉硫磷質量濃度呈線性關系,線性回歸方程為Y=0.930 07X+2.695 75×10-4,R2=0.995,檢出限為45.54 ng/mL。在馬拉硫磷質量濃度為300 ng/mL時,AuNPs已經開始變色,為防止視覺誤差,馬拉硫磷目測檢出限為400 ng/mL。本方法與其他檢測方法的比較如表1所示,國標中規定的生菜和蘋果中馬拉硫磷的最大殘留限量最低為500 ng/mL,本方法的檢出限遠低于國標限量,且在較大的線性范圍內均適用。
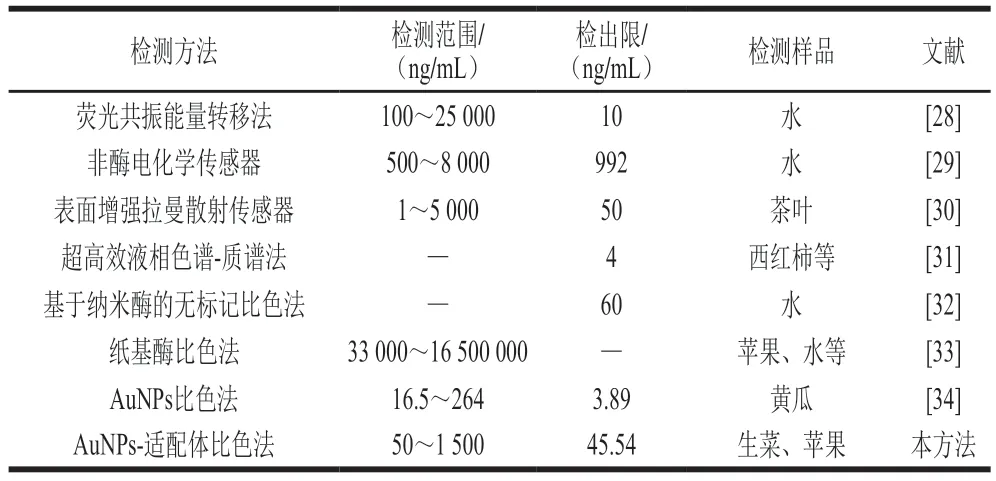
表1 文獻報道方法與本法檢測馬拉硫磷的比較Table 1 Comparison of this method and other reported ones for the determination of malathion
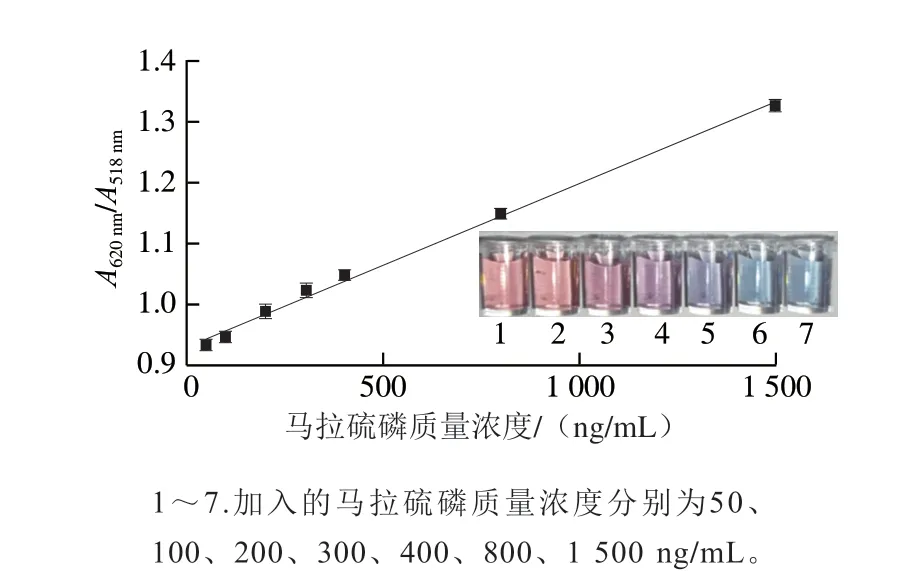
圖10 不同馬拉硫磷質量濃度下A620 nm/A518 nm值Fig.10 A620 nm/A518 nm ratio at different malathion concentrations
2.4 特異性鑒定
本研究選擇馬拉硫磷、樂果、乙酰甲胺磷、毒死蜱、敵敵畏、草甘膦、吡蟲啉、倍硫磷考察方法的特異性,如圖11所示,在體系中加入800 ng/mL的馬拉硫磷、樂果、乙酰甲胺磷、毒死蜱、敵敵畏、草甘膦、吡蟲啉、倍硫磷,除馬拉硫磷外,加入其他農藥時A620nm/A518nm值較低,與陰性對照大致相同,AuNPs不發生聚集;而加入馬拉硫磷時,體系中A620nm/A518nm值較大,AuNPs發生聚集現象。表明本方法對馬拉硫磷具有特異性識別能力,且馬拉硫磷結構類似物及其他有機磷農藥不會影響馬拉硫磷殘留的特異性檢測。
2.5 實際樣品檢測結果
為驗證建立的AuNPs-適配體比色傳感法的準確性和可靠性,采用本方法對生菜和蘋果樣品進行檢測。從本地超市購得生菜和蘋果樣品中未檢出馬拉硫磷。通過向預處理的樣品中添加馬拉硫磷標準品制成不同質量濃度(200、400、800 ng/mL)的加標檢測液。使用本方法進行檢測,每個樣品重復測定3 次。如表2所示,基于AuNPs-適配體的比色傳感法檢測馬拉硫磷的加標回收率范圍分別為98.24%~100.91%和99.48%~101.29%,相對標準偏差分別為0.2%~2.2%和0.2%~2.8%。上述結果表明,基于適配體建立的AuNPs-適配體比色傳感法具有良好的準確性和可靠性。本方法可用于生菜和蘋果中馬拉硫磷的快速篩查和現場檢測。
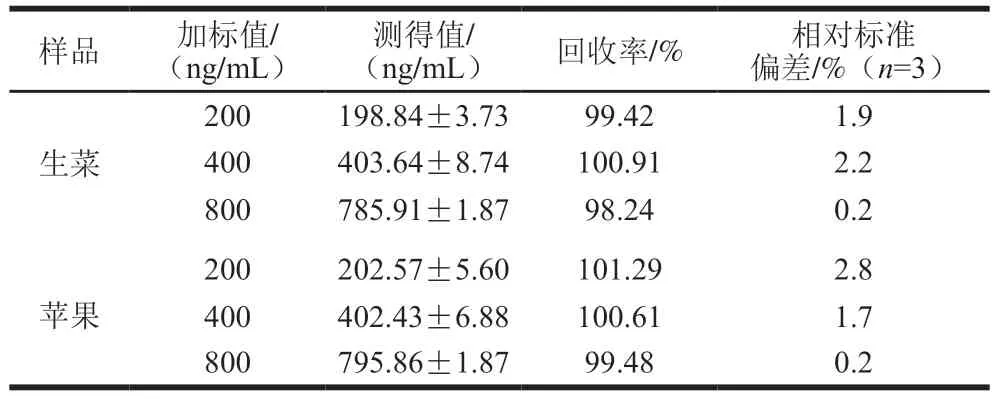
表2 馬拉硫磷在生菜和蘋果中的加標回收結果(本方法)Table 2 Recoveries of malathion from spiked lettuce and apple samples using this method
2.6 方法驗證結果
國標法檢測結果如表3所示,將本方法的檢測結果與國標法的結果進行對比(圖12),結果顯示兩者的準確度一致,生菜樣品和蘋果樣品的R2均為0.999,證實了本方法的可靠性。
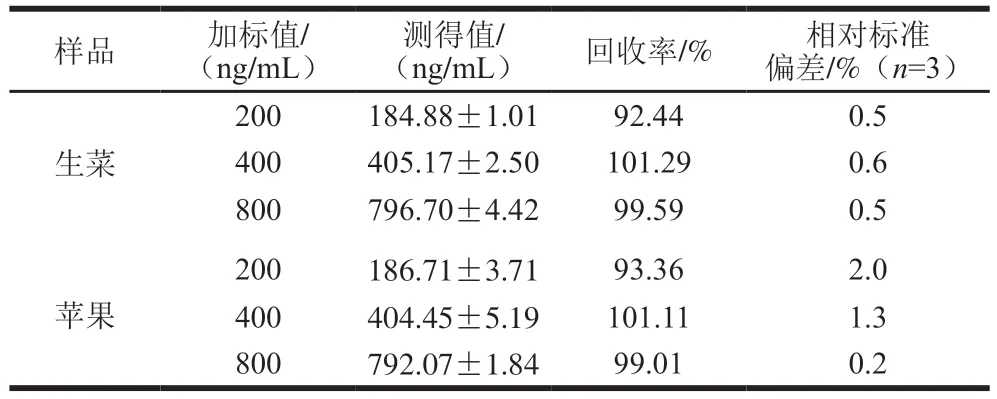
表3 馬拉硫磷在生菜和蘋果中的加標回收結果(國標法)Table 3 Recoveries of malathion from spiked lettuce and apple samples using the national standard method
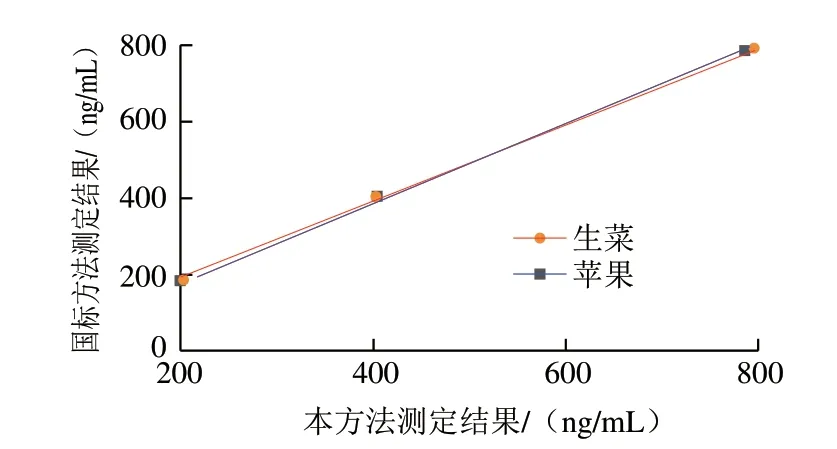
圖12 本方法和國標法的加標回收結果對比Fig.12 Comparison of recoveries of this method with those of the national standard method
3 結 論
以AuNPs為指示劑,馬拉硫磷適配體作為識別元件,建立了AuNPs-適配體比色傳感法。通過優化溶液濃度,最后得到NaCl最適濃度為500 mmol/L,適配體濃度為1 μmol/L。通過優化孵育時間,最終得到的NaCl與AuNPs最佳反應時間為10 min,馬拉硫磷適配體與AuNPs最佳反應時間為10 min。本方法在馬拉硫磷質量濃度為50~1 500 ng/mL范圍內呈現良好的線性關系,線性相關系數R2=0.995,根據計算得到本方法檢出限為45.54 ng/mL,
目測檢出限為400 ng/mL。本方法簡單快速,易于進行定性和定量分析,具有優異的靈敏性、特異性和準確性。經驗證,本方法在實際加標樣品中的檢測結果與國標法的檢測結果高度一致,是一種可行的快速檢測食品中馬拉硫磷殘留的高效方法,可為其他農藥的現場快速篩查方法研究提供參考。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
