自噬對肝衰竭的保護作用機制與臨床價值
2023-11-06 09:01:06胡洋洋張興羅越王亞東趙彩彥
臨床肝膽病雜志 2023年10期
胡洋洋, 張興, 羅越, 王亞東, 趙彩彥
河北醫科大學第三醫院湘江院區感染肝病科, 石家莊 050051
肝衰竭是由多種因素引起的肝臟嚴重損害,具有病情進展迅速、病死率高等特點。以免疫炎癥損傷為核心的“二次打擊學說”是肝衰竭發病關鍵機制。細胞自噬在肝臟免疫炎癥損傷過程中發揮關鍵調控作用。近年研究表明自噬通過調節炎癥小體活化、對抗氧化應激、抑制細胞凋亡等機制對肝衰竭發揮保護效應。本文就自噬在肝衰竭中的變化、調控機制及其作為肝衰竭治療靶點的潛在前景作一綜述。
1 自噬在肝衰竭中的變化特點
自噬是一種進化上高度保守的細胞適應性保護機制,由30余種自噬相關基因(autophagy related gene,Atg)及其編碼的Atg蛋白分子家族所介導,參與細胞質質量控制、新陳代謝、免疫調控等過程。尤其選擇性自噬通過降解受損細胞器,發揮細胞“清道夫”作用[1]。在肝衰竭的發生發展過程中存在自噬受損現象,而代償性激活自噬可通過抑制肝細胞內蛋白質聚集、脂質堆積、氧化應激、細胞炎癥與死亡,維持肝細胞生存穩態。
Ren等[2]研究證實,HBV相關急性肝衰竭(acute liver failure,ALF)患者肝組織自噬相關分子Atg7、Atg5、Beclin-1表達顯著降低。同樣,慢加急性肝衰竭(acute-on-chronic liver failure,ACLF)患者肝組織Atg5、Beclin-1、微管相關蛋白1輕鏈3(microtubule associated protein 1 light chain 3,LC3)-Ⅱ/LC3-Ⅰ的表達也顯著降低,且伴隨自噬底物連接蛋白p62聚集性升高[3]。Wu等[4]研究則發現終末期肝病模型(model for end-stage liver disease,MELD)評分>25的ACLF患者脂多糖(lipopolysaccharide,LPS)水平顯著高于MELD評分≤25患者,進一步體外實驗顯示LPS通過p38MAPK信號通路抑制肝星狀細胞(hepatic stellate cell,HSC)自噬,并伴隨HSC來源的IL-1β及其前體表達增加。上述研究證實肝衰竭患者存在自噬受損并可因此誘發或加重肝臟炎癥反應。由D-氨基半乳糖(D-galactosamine,D-GalN)/LPS或CCl4誘導的肝衰竭動物模型顯示,在急性肝損傷(acute liver injury,ALI)早中期,肝臟自噬水平升高,當進展至肝衰竭階段自噬水平卻降低,激活自噬顯著減輕肝臟炎癥損傷,提高ALF小鼠存活率[2,5]。因此,自噬在肝衰竭發生發展不同階段存在動態變化,且對肝衰竭發揮保護作用。在對乙酰氨基酚(acetaminophen,APAP)誘導的動物和肝細胞損傷模型中也均可觀察到自噬激活現象,后者通過去除受損線粒體發揮肝臟保護作用[6-7]。但過量APAP通過損害自噬通量,導致受損線粒體不能被有效清除,增加線粒體氧化應激,造成肝細胞壞死[8]。值得注意的是,刀豆素A誘導的ALI小鼠模型中,自噬呈過度激活并誘導樹突狀細胞活化,加劇肝臟免疫炎癥反應,甚至觸發肝細胞自噬性死亡,小鼠存活率顯著降低[9]。由此可見,雖然不同病因、不同階段的肝衰竭存在自噬強度和方向的差異,也影響著肝衰竭疾病轉歸和結局,但更多證據支持適度自噬有利于限制肝衰竭發生發展。
2 自噬參與肝衰竭保護作用的機制
2.1 調節Nod樣受體熱蛋白結構域相關蛋白3(Nod-like receptor pyrin domain-containing protein 3,NLRP3)炎癥小體信號 NLRP3炎癥小體由Nod樣受體、凋亡相關斑點樣蛋白(apoptosis-associated speck-like protein containing a CARD,ASC)和半胱氨酸天冬氨酸蛋白酶-1(cysteiny aspartate-specific protease-1,caspase-1)組成,通過介導IL-1β和IL-18成熟信號,調節炎癥和免疫反應。Jia等[10]發現HBV相關ACLF患者肝組織中NLRP3及caspase-1、IL-18和IL-1β表達水平升高,是造成肝臟持續性炎癥損傷的重要原因。ALF小鼠模型肝組織中NLRP3、ASC、caspase-1等也顯著升高,并伴隨肝細胞空泡變性和壞死增加[11],因此NLRP3炎性小體激活是肝衰竭炎癥損傷核心機制之一。
在D-GalN/LPS誘導的ALF小鼠模型中,應用組蛋白去乙酰化酶抑制劑CAY10683修飾自噬相關蛋白ULK1 K68賴氨酸位點,上調ULK1表達,繼而抑制NLRP3活化改善肝功能[12]。應用LPS處理人HSC細胞株LX2后,Atg13、LC3-Ⅱ表達呈劑量依賴性下降,并伴隨NLRP3、IL-1β及其前體的表達增加,當LX2細胞暴露于自噬抑制劑巴佛洛霉素A1可進一步促進p62積累、NLRP3活化和IL-1β產生[4]。上述研究均提示自噬參與對NLRP3炎性小體的調控,并因此影響肝臟免疫炎癥損傷。此外,NF-κB參與上調NLRP3和IL-1β前體等轉錄表達,是NLRP3炎癥小體激活的重要信號。Shan等[13]發現APAP誘導的ALI小鼠肝組織NLRP3、caspase-1、IL-1β表達均明顯增加,自噬激活劑雷帕霉素(rapamycin,RAPA)可顯著抑制APAP誘導的NF-κB核移位以及NLRP3通路活化。反之,抑制自噬則使IκBα受抑和p-NF-κBp65蛋白表達增加,激活NF-κB,上調NLRP3信號通路相關炎癥因子的轉錄和翻譯。
體外細胞研究[14]也證實,生長停滯特異蛋白6信號通路介導的自噬通過抑制肝臟Kupffer細胞中caspase-1的激活,抑制促炎因子生成,阻止肝臟炎癥反應進展。此外,應用LPS刺激體外培養的單核細胞顯示ASC呈p62依賴性降解,抑制炎癥小體活性[15]。綜上,自噬通過抑制NF-κB核移位減少炎癥小體轉錄、抑制caspase-1活性以及吞噬和降解NLRP3炎性小體成分等機制負性調控NLRP3炎癥小體活性,從而減輕肝衰竭炎癥損傷。
2.2 抑制氧化應激 氧化應激介導的肝損傷也是肝衰竭發病機制的重要環節,由于機體氧化與抗氧化系統失衡,活性氧(reactive oxygen species,ROS)過度生成和積聚,從而造成肝細胞廣泛而不可逆性死亡。在LPS/D-GalN誘導的ALF小鼠肝組織以及LPS處理的巨噬細胞RAW264.7和HSC中均可檢測到大量ROS產生,從而消耗內源性抗氧化物質,導致核酸、蛋白質、脂質等細胞成分受損,加劇肝細胞死亡[16-17]。
研究發現,線粒體自噬通過選擇性清除受損線粒體,減少APAP誘導的受損肝細胞ROS產生,減輕肝臟氧化應激性損傷。在自噬相關分子Atg5缺陷的敗血癥小鼠模型中,肝細胞內受損線粒體大量積累,通過增加線粒體ROS的產生并啟動線粒體凋亡途徑,加速器官功能衰竭[18]。自噬選擇性清除功能障礙線粒體的機制復雜,可能與PTEN誘導性激酶蛋白1(PTEN induced putative kinase 1,PINK1)-Parkin信號通路相關。Wang等[19]通過電鏡觀察發現PINK1和Parkin雙敲除小鼠在APAP處理后自噬小體和溶酶體數量減少,并且線粒體蛋白泛素化和p62蛋白移位明顯減少,延遲肝臟谷胱甘肽還原,進而加速肝細胞壞死及小鼠死亡。自噬調節氧化應激的另一機制由核因子E2相關因子2(nuclear factor erythroid 2-related factor,Nrf2)介導。生理條件下,Nrf2與Kelch樣ECH相關蛋白1結合而處于失活狀態,當發生氧化應激時可被磷酸化的p62激活,繼而結合細胞核抗氧化反應元件,上調抗氧化酶基因轉錄,參與細胞對氧化應激性損傷的防御。在過量APAP誘導的ALI早期,p62、磷酸化p62水平顯著上調,并促進Nrf2向細胞核內移位,上調抗氧化基因血紅素加氧酶-1、谷氨酸-半胱氨酸連接酶催化亞基(glutamate-cysteine ligase catalytic subunit,GCLC)等細胞保護酶表達,代償性對抗氧化應激,下調肝細胞ROS水平,降低肝細胞損傷風險[20]。Ruart等[21]檢測CCl4處理的大鼠肝竇內皮細胞顯示早期階段(4周)可觀察到自噬增強,而晚期(6周)自噬強度不再變化。進一步通過敲除肝竇內皮細胞Atg7的動物模型證實,僅在肝竇內皮細胞中Nrf2依賴性抗氧化基因GCLC、谷胱甘肽-S-轉移酶Mu等表達上調,提示自噬缺陷是Nrf2激活的重要觸發因素。綜上,在肝衰竭發病機制中自噬與Nrf2信號通路均參與調控氧化應激與抗氧化機制的平衡,并且在其調控過程中呈現時間和空間差異。
自噬對抗氧化應激的過程同時也參與NLRP3炎癥小體活性調控,說明兩種機制存在交叉。高濃度ROS可使硫氧還蛋白相互作用蛋白與硫氧還蛋白解離,直接與NLRP3相互作用,激活NLRP3炎癥小體。Yang等[22]檢測RAPA處理的肝損傷小鼠模型肝組織顯示,增強的自噬能夠清除過量釋放的ROS,下調硫氧還蛋白相互作用蛋白/NLRP3軸,從而抑制小鼠肝臟中NLRP3炎癥小體活化。由PINK1介導的線粒體自噬也可通過清除線粒體ROS抑制肝組織NLRP3炎癥小體活化、線粒體氧化應激,從而減輕黃曲霉素B1誘導的小鼠肝組織炎癥損傷[23]。上述研究揭示了自噬和氧化應激、NLRP3炎癥小體之間的新聯系,自噬在肝衰竭發病機制的調控中具有樞紐作用。
2.3 調控細胞凋亡 TNF/TNF受體系統激活介導的細胞凋亡同樣是ALF發病重要分子機制之一。自噬調控細胞凋亡對維持細胞生存具有重要意義。Ahmedy等[24]在LPS/D-GalN誘導的ALF小鼠肝組織中觀察到細胞凋亡增加,p53和caspase-3、Bax表達顯著上調,而在誘導ALF發生前應用柚皮素可使升高的caspase-3、p53和Bax水平分別降低49%、34%和54%,并且這種抑制細胞凋亡的作用可被自噬抑制劑3-甲基腺嘌呤消除。不僅如此,自噬還可在轉錄水平抑制TNF-α等炎癥因子表達,或抑制TNF-α介導的半胱氨酸蛋白酶激活途徑保護肝細胞免于凋亡。另一方面,線粒體膜電位下降是細胞凋亡的早期事件,自噬抑制劑3-甲基腺嘌呤通過降低ALF體外模型細胞L-02線粒體膜電位,增加細胞凋亡相關蛋白Bax、細胞色素C水平,對抗自噬介導的抗肝細胞凋亡作用[25]。Oami等[18]通過透射電鏡觀察發現,條件性Atg5基因敲除小鼠的肝細胞線粒體損傷和細胞凋亡加速,可能由于自噬缺陷誘導了線粒體死亡途徑,促進肝細胞凋亡。自噬不僅參與調控肝細胞凋亡,Tian等[26]發現抑制自噬也可以促進HSC凋亡,導致細胞外基質生成減少,加速ALF肝臟實質結構的塌陷;利用RAPA激活自噬可抑制一氧化氮介導的細胞凋亡,保護HSC,為再生肝細胞提供支架。以上研究表明,自噬參與肝細胞和HSC凋亡的調控過程以減輕肝組織損傷,并且TNF-α、線粒體膜電位、一氧化氮等分子信號在其中發揮重要作用(圖1)。
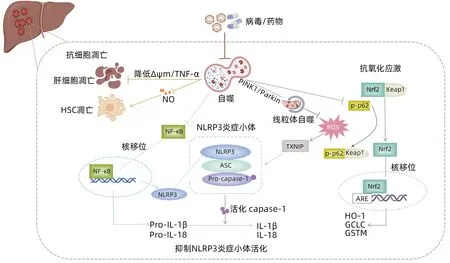
圖1 自噬在肝衰竭中的保護作用機制Figure 1 The protective mechanism of autophagy in liver failure
3 調控自噬對肝衰竭治療的價值
3.1 外泌體與自噬 外泌體是膜衍生的納米級囊泡(直徑為30~150 nm),可攜帶蛋白質、脂類和核酸等多種組分,參與清除致病或受損的細胞內物質、介導細胞間通訊[27-28]。越來越多研究[29]證實,自噬與外泌體存在交互作用,自噬可以降解功能失調的外泌體,而外泌體又可以通過誘導細胞自噬發揮細胞保護作用。Shen等[30]建立Atg5-/-AML12細胞模型顯示,經IL-1β/TNF處理后的AML12細胞可分離出含損傷相關分子模式的外泌體,后者進一步上調Kupffer細胞IL-1、IL-6等基因表達,導致細胞能量穩態失衡、溶酶體通透改變,加重炎癥反應。國內學者最新臨床研究[31-33]證實肝衰竭患者肝臟來源的外泌體數量、組分及功能方面均存在差異性變化,且影響著肝衰竭患者疾病嚴重程度和預后轉歸。
Yang等[34]體外誘導骨髓間充質干細胞分化為肝細胞樣細胞后分離提取外泌體,并將其應用于肝臟缺血再灌注小鼠模型顯示,處理后小鼠肝組織LC3-Ⅱ、Beclin-1水平升高,p62/SQSTM1水平降低,抑制肝細胞變性和壞死,證實外泌體可以通過增強自噬減輕肝臟損傷。此外,人骨髓間充質干細胞來源外泌體通過輸送微小核糖核酸let-7a-5p靶向MAP4K3-轉錄因子EB信號通路恢復ACLF小鼠肝組織自噬通量,有效減輕肝細胞損傷和凋亡[35]。綜上,自噬缺陷可能促進肝細胞分泌損傷相關分子模式外泌體,激活巨噬細胞引起炎癥,而干細胞來源的外泌體則通過增強自噬發揮肝臟保護作用,為臨床應用外泌體治療肝衰竭提供策略。
3.2 過氧化物酶體增殖物激活受體α(PPARα)與自噬 PPARα激動劑亦可通過促進ALF小鼠Atg5、Atg7、溶酶體膜相關蛋白-1和LC3-Ⅱ表達,延緩ALF進展;抑制自噬則逆轉PPARα的肝臟保護作用,促進炎癥因子和趨化因子表達上調,加劇肝臟炎癥損傷。Ren等[2]證實抑制糖原合成酶3β(glycogen synthase kinase 3β,GSK3β)活性可通過增加PPARα表達激活自噬減輕肝臟損傷,應用siRNA沉默PPARα表達則逆轉上述作用。磷酸腺苷活化蛋白激酶(AMP-activated protein kinase,AMPK)也通過影響PPARα活性參與肝衰竭自噬激活。Liu等[36]報道在miR-19a調控的肝細胞AMPK/PPARα自噬信號通路中,miR-19a通過激活AMPK增強PPARα的轉錄活性上調LC3-Ⅱ和Beclin-1表達。此外,PPARα激動劑非諾貝特通過激活自噬、降低肝臟ROS和脂質過氧化有效減輕APAP誘導的ALI。因此,調控PPARα活性為靶點的信號分子可為激活自噬發揮肝衰竭保護作用提供新思路。
3.3 其他 自噬作為拮抗肝細胞炎癥和壞死的核心機制,還受其他多種分子信號調控。例如,RAPA靶蛋白抑制劑西羅莫司可增強自噬,減輕肝細胞損傷與死亡,目前已在動物或體外實驗模型中應用[22,37]。煙酰胺磷酸核糖轉移酶抑制劑FK866通過抑制c-Jun氨基末端激酶信號通路誘導自噬,對D-GalN/LPS和刀豆素A誘導的ALF小鼠發揮保護作用[38]。可溶性T細胞免疫球蛋白黏蛋白分子3也可以增加ALF小鼠肝臟CD11b+巨噬細胞中LC3-Ⅱ的水平,促進自噬小體形成,減少肝臟炎癥介質產生[39]。骨髓間充質干細胞也可以通過PI3K/Akt途徑調節自噬以減輕ALF肝細胞凋亡與肝組織炎癥反應,而其旁分泌的血紅素加氧酶-1也參與自噬的調節[40]。雖然上述研究尚有待進一步深入,但相關機制探索為臨床發掘基于自噬調控的分子靶向治療提供了一系列的研究思路和開發靶位。
4 結語與展望
綜上,自噬是保護肝細胞免受炎癥損傷的重要機制。靶向激活自噬將可能成為肝衰竭治療領域的重要課題。雖然目前將調控自噬作為肝衰竭治療的靶點尚存在諸多問題和體內實踐應用的風險,包括:(1)目前研究大多局限于細胞和動物模型,在肝衰竭特定的炎癥背景下激活自噬的具體分子調控機制和信號通路仍待深入;(2)自噬信號通路涉及多種分子調節機制,但目前尚未評價出最佳分子調控靶位;(3)尚無特異性靶向肝臟的自噬激活劑,在激活肝臟自噬的同時可能使患者面臨其他臟器細胞自噬性死亡和腫瘤發生的風險;(4)自噬具有抑制HSC凋亡為再生細胞提供支架作用,但HSC凋亡過度抑制可能導致肝纖維化加重,降低自噬激活劑在臨床應用的安全性。但自噬通過參與調控NLRP3炎癥小體介導的炎癥反應、對抗氧化應激、抑制細胞凋亡等方式發揮對肝衰竭的保護作用毋庸置疑,以調控外泌體、PPARα等為靶點的自噬活化策略也將有望成為肝衰竭分子靶向治療的重要策略。
利益沖突聲明:本文不存在任何利益沖突。
作者貢獻聲明:胡洋洋負責查閱文獻,撰寫文章;張興、羅越負責校對文章;王亞東負責指導立題及修改;趙彩彥負責審閱文章等。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
學苑創造·A版(2020年9期)2020-10-13 09:41:02
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
西南軍醫(2016年6期)2016-01-23 02:21:19
新疆醫科大學學報(2015年10期)2015-12-26 12:33:30
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
西南軍醫(2015年2期)2015-01-22 09:09:37
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
食品科學(2013年15期)2013-03-11 18:25:48
