腦腎素-血管緊張素系統通過腦微血管內皮細胞調控神經血管單元介導阿爾茨海默病機制研究進展
2023-11-06 07:31:18張評奧宋成寰施霽耘黃婉瑩張永芳
中國藥理學與毒理學雜志 2023年10期
關鍵詞:氧化應激
張評奧,宋成寰,施霽耘,黃婉瑩,張 瑞,張永芳,王 昊
(上海交通大學醫學院1.藥理學與化學生物學系,2.上海高校轉化醫學協同創新中心,上海 200025)
阿爾茨海默病(Alzheimer disease,AD)是全球最常見的神經退行性疾病,其發病率逐年上升,至2050 年,全世界發病率相較于2021 年或將增長3 倍[1]。AD 的主要病理特征為海馬和皮質區的細胞外β-淀粉樣蛋白(amyloid β-protein,Aβ)沉積、細胞內神經原纖維纏結(neurofibrillary tangle,NFT)以及神經炎癥[1]。AD患者常表現出明顯的記憶、語言和情感障礙及空間識別和學習能力下降[1]。值得注意的是,腦血管健康失衡是所有神經退行性疾病的共同病理特征之一,在AD 患者中普遍存在。AD患者還常伴隨血壓升高、血管狹窄和腦血流量減少等癥狀,同時會出現微血管堵塞、出血、異常血管增生和血管基底層丟失等病理改變[2]。AD 的血管病理學說認為,早期AD 是一種微血管疾病,腦血管功能異常是AD 最早出現的生物學特征之一[2]。
腦微血管內皮細胞(brain microvascular endothelial cell,BMEC)是構建腦毛細血管和組成血腦屏障(blood brain barrier,BBB)的基本細胞,同時也是神經血管單元(neurovascular unit,NVU)的重要成員[3]。腦微血管滲透壓及生理功能依賴腎素-血管緊張素系統(renin-angiotensin system,RAS)的調節,而以血管緊張素Ⅱ(angiotensinⅡ,AngⅡ)水平升高為主的RAS失衡則是誘導AD血管損傷的重要誘因[4]。現已證實,腦組織中的各類細胞可分別獨立地表達RAS 中的各個組分,其獨立于外周RAS 在腦組織形成一套特殊的局部RAS——腦RAS(brain RAS,BRAS)。BRAS 的主要成分包括星形膠質細胞分泌的血管緊張素原(angiotensinogen,AGT)、神經元細胞與小膠質細胞表達的腎素和血管緊張素受體(angiotensin receptor,ATR)及腦微血管內皮細胞表達的血管緊張素轉換酶(angiotensin converting enzyme,ACE)(圖1)。BRAS 產生的AngⅡ并非來自循環而是直接在腦組織中產生,不作用于外周,而是直接作用于鄰近的神經細胞,因此相比于外周RAS,BRAS 對維持中樞神經細胞功能具有更重要的作用[4-5]。而BMEC作為外周RAS 和BRAS 共同的效應細胞,可能是實現RAS 與BRAS 調控的重要橋梁。盡管RAS 在外周循環系統疾病中的作用已被深入研究,但其對神經細胞的調控作用并未獲得廣泛關注,其調節機制也尚未明確。研究表明,BRAS 作為局部調控系統在調控NVU 和AD 的發生發展中發揮了重要作用[5]。本文擬圍繞BRAS 介導的NVU 調控網絡及其與AD關系的研究進展進行綜述。
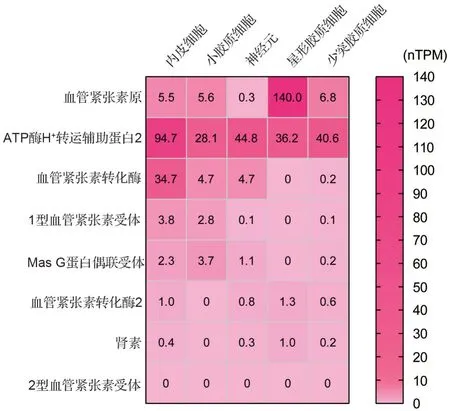
圖1 人腦中樞神經系統(CNS)主要細胞中腎素-血管緊張素系統(RAS)關鍵成員RNA 表達與分布. nTPM:每百萬堿基的轉錄本(transcripts per kilobase million).數據獲取自THE HUMAN PROTEIN ALTAS(https://www.proteinatlas.org/)中的scRNA-seq數據庫.
1 BRAS參與AD發生發展
BRAS 的運轉機制與外周RAS 相似,在腦星形膠質細胞中合成的AGT 會被中樞游離的腎素代謝為AngⅠ,隨后在BMEC、神經元和膠質細胞的細胞膜上被ACE 催化代謝為AngⅡ。AngⅡ可通過1 型血管緊張素受體(angiotensin type 1 receptor,AT1R)調節細胞的生理功能。AngⅡ隨后被ACE2 轉化代謝為Ang(1-7),而激活Mas 受體(Masreceptor,MasR)可對AngⅡ產生拮抗作用(圖2)[4-5]。
AngⅡ具有強烈的縮血管活性,病理狀態下,AngⅡ具有較強的促炎和促氧化作用[4-5]。AngⅡ的異常升高是高血壓等心血管疾病的特征指標,也是血管性癡呆的重要誘因[6]。值得注意的是,AngⅡ是相對分子質量為1046 的非脂溶性多肽類化合物,由于其親水特性,外周AngⅡ難以透過BBB[7],因此,BRAS 對微環境穩態具有獨立于循環系統RAS 的重要調控作用。臨床研究表明,AD 患者腦組織中AGT 和AngⅡ顯著升高,而腦脊液和血清中的ACE 蛋白水平和活性降低,內側額葉皮質中ACE2 活性也顯著降低[5,8-9]。ACE 是水解AGT 的關鍵酶,已有研究證實,ACE 具有水解Aβ1-42和抑制Aβ 沉積的作用[10]。注射外源性AngⅡ會造成血管功能損傷,促進Tg2576 小鼠Aβ 沉積,加劇認知損傷[11]。AngⅡ可通過激活AT1R 增加淀粉樣前體蛋白mRNA 水平,或通過增強β-分泌酶活性和增加早老素表達等多種途徑提高腦內Aβ 水平[12]。另有研究發現,AngⅡ與微管蛋白過度磷酸化、氧化應激和神經炎癥有關[12-13]。衰老大腦表現出AT1R 上調及AT2R 下調的生理特征也間接提示AngⅡ在神經退行性疾病中的重要作用[14]。總之,BRAS 失調,特別是AngⅡ水平升高,可通過氧化應激和神經炎癥等機制參與AD的發生發展(圖2)。
迄今為止,誘導AD 患者BRAS 失衡的確切機制尚不明確,有研究提示,BRAS失衡與血管病變密切相關,其中AT1R 過度激活與腦血管缺血、腦氧化應激、BBB 破壞和炎癥等密切相關[4]。ACE2 表達和活性的降低則與高血壓、心力衰竭、動脈粥樣硬化和糖尿病等血管相關疾病有關[15]。研究表明,高血壓期間循環系統AngⅡ水平升高會破壞BBB 完整性,從而使外周循環系統中的AngⅡ能夠進入交感神經中樞,誘導神經炎癥[7]。簡言之,維護血管健康,同時維持BRAS 穩態對預防AD 的發生及抑制其發展均具有重要意義。目前已開展多項RAS 藥物改善認知障礙的臨床試驗,其中AT1R 拮抗劑(沙坦類藥物)和ACE 抑制劑(普利類藥物)具有良好的BBB 透過性,多項Ⅱ期臨床試驗結果顯示,抑制BRAS具有改善認知功能的效應[16-20]。相比于其他RAS 抑制劑,沙坦類藥物尤其是坎地沙坦具有更好的改善患者認知障礙的作用,且對患者血壓和腦血流影響更小,更具開發潛力(表1)[16-31]。
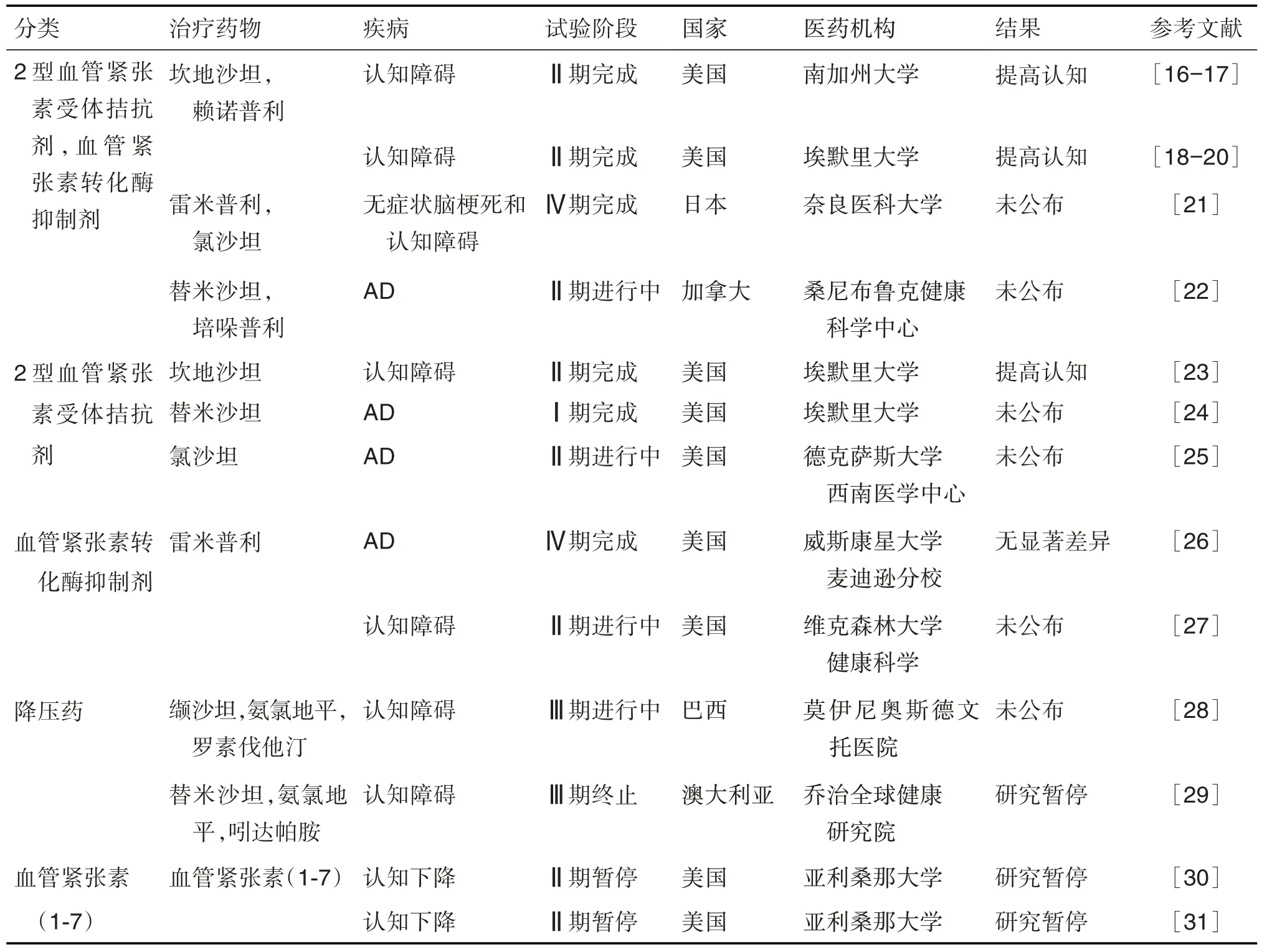
表1 抗RAS藥物改善認知的臨床試驗
2 NVU調節網絡與AD發病機制
AD 發病機制的血管病理假說認為,腦血管系統通過能量傳輸、氧氣運輸和傳遞信號分子等途徑維持中樞神經系統(central nervous system,CNS)微環境穩態,神經細胞也可通過信號轉導調節血管狀態,以滿足CNS的物質與能量代謝需求。神經與腦血管間的這種交互網絡被稱為神經血管耦合(neurovascular coupling,NVC),其功能單位稱為NVU[3]。NVU 主要由BMEC、膠質細胞和神經元組成,NVU 同樣是構成BBB 的物質基礎,NVU 成分間借助NVC 相互作用,并影響BBB 的結構完整性和功能[32]。NVU 功能失調可引起腦缺氧和BBB 破壞,導致外周毒素入腦,促進炎癥和氧化應激產生,同時也會影響Aβ 清除,加速中樞Aβ 積累和NFT 形成,進一步惡化血管功能障礙,進入惡性循環,加重AD 病理[33-34]。神經元、腦微血管內皮細胞、星形膠質細胞和小膠質細胞中任一成員的病理變化均可通過NVU調控網絡進入惡性循環,破壞中樞微環境穩態。如小膠質細胞激活后可釋放炎癥因子,誘導反應性星形膠質細胞激活和異常增生、星形膠質細胞終足脫落并釋放趨化因子破壞BBB 及外周毒素入腦并損傷神經元,最終導致神經退行性病變(圖3)。
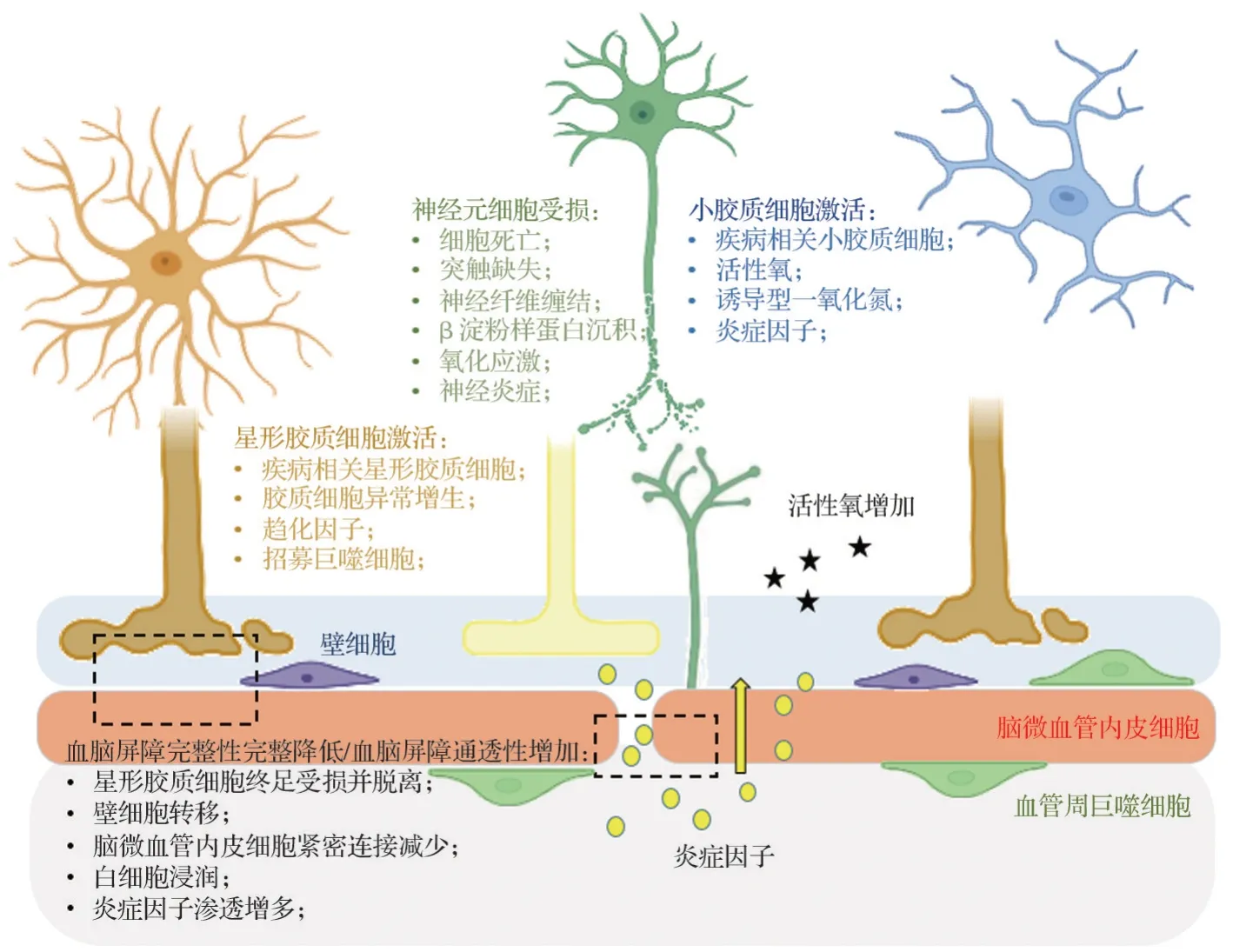
圖3 神經血管單元(NVU)損傷誘導AD 發生. 病理狀態下NVU 各成員細胞表現中出現如星形膠質細胞異常增生、神經元死亡、血管內皮細胞緊密連接降低、壁細胞遷移和小膠質細胞活化等病理學改變,破壞中樞神經系統微環境穩態,導致細胞氧化應激、神經炎癥和代謝異常的發生,并最終誘導AD發生.
3 BRAS對NVU的調節作用
NVU 的調節網絡十分復雜,其內部機制尚未完全闡明,而BRAS 的主要組件分別位于腦神經元和神經膠質細胞,AT1R 則幾乎分布在所有血管和神經細胞上,因此AngⅡ可能在NVU 幾種關鍵細胞間的信號傳導和交流互作方面發揮調節作用,具有調節NVU 的作用[35]。CNS 中,星形膠質細胞通過表達AGT 調節AngⅡ水平,后者通過AT1R 作用于BMEC、神經元和神經膠質細胞,進而調節BBB 通透性和CNS微環境穩態。由此可見,BRAS是NVU調節網絡中的重要環節,而AGT/AngⅡ/AT1R 則是NVU成員間實現交互串擾的重要途徑。
3.1 BRAS對BMEC的調節作用
BMEC是AngⅡ重要的靶細胞。除了調節血管的緊張狀態,AngⅡ也可通過凋亡信號調節激酶1和內皮素介導BMEC 損傷,并通過激活AT1R 破壞BMEC 間的緊密連接,降低電阻,從而提高BBB 經細胞和旁細胞通透性,破壞BBB 的完整性和屏障功能[36-37]。AngⅡ損傷BMEC 通透性的機制可能與AT1R 介導的過氧化物酶體增殖物激活受體α 去磷酸化有關[38]。近期Ding[39]等發現,AngⅡ可通過NADPH 氧化酶2 促進BMEC 中超氧化物的產生。此外,RAS 的激活也會抑制血管內皮細胞一氧化氮合酶磷酸化,從而影響一氧化氮(nitric oxide,NO)合成與釋放,介導內皮細胞功能障礙,進而抑制細胞遷移[39-40]。
3.2 BRAS對神經元的調節作用
研究發現,AngⅡ可直接作用于神經元,并影響其電信號傳遞、氧化應激和炎癥反應[41]。Case等[42]認為,AngⅡ介導的神經元損傷由線粒體超氧化物的增加所致,并提出神經元線粒體中NADPH氧化酶4 會通過產生超氧化物促進AngⅡ介導的神經元損傷。Ma 等[43]研究表明,AngⅡ可通過促進突觸處的鈣通道亞基α2δ-1 與N-甲基-D-天冬氨酸受體結合,增強交感神經元興奮性。Garrido-Gil等[44]則發現,AngⅡ通過AT1R 在多巴胺能神經元鐵穩態調節中發揮重要作用。激活AT2R 或Ang(1-7)-MasR 軸可改善認知障礙,并改善AngⅡ對神經元的損傷作用[45-46]。Kim 等[47]發現,RAS 抑制劑具有顯著的神經保護作用,可糾正帕金森病模型斑馬魚的代謝異常。因此有研究指出,抑制AT1R 激活的同時選擇性激活AT2R 是改善AD 中神經元功能障礙的潛在策略[46]。
3.3 BRAS對星形膠質細胞的調節作用
研究發現,AngⅡ可誘導星形膠質細胞炎癥和氧化應激,并抑制星形膠質細胞對谷氨酸的攝取[48-49]。Diaz 等[50]和Boily 等[51]發現,AngⅡ增加星形膠質細胞上瞬時電位辣椒素受體4通道蛋白的表達,提高細胞內Ca2+濃度和相關信號強度,并引起星形膠質細胞終足與血管內皮細胞間的緊密連接受損。Nataf等[52]認為,AngⅡ是星形膠質細胞中水通道蛋白4 與轉化生長因子β 間的信號傳遞介質。值得注意的是,星形膠質細胞是CNS 中AGT的主要來源,當敲除AGT基因后會導致星形膠質細胞終足從血管內皮細胞上脫離,并引起BBB 受損;補充外源性AngⅡ則有助于緩解這種損傷,而應激狀態下產生的AGT則會加劇神經炎癥[53-54]。AngⅡ不僅參與調控星形膠質細胞活化、炎癥激活和氧化應激,也可通過星形膠質細胞調節BBB 功能或介導神經元損傷。
3.4 BRAS對小膠質細胞的調節作用
小膠質細胞極化可通過吞噬神經元突觸介導突觸損失,加劇微管蛋白磷酸化,并分泌炎癥因子,或通過激活星形膠質細胞加劇對神經元的損傷[55]。研究發現,BRAS 與雌激素、Rho 激酶、胰島素樣生長因子1、腫瘤壞死因子α、鐵離子、過氧化物酶體增殖物激活受體γ 和Toll樣受體等多種小膠質細胞的極化因子間存在關聯[14,44,56]。AngⅡ可激活小膠質細胞Toll 樣受體4/MYD88 先天免疫信號轉導適配器/NF-κB 信號通路誘導小膠質細胞活化,活化的小膠質細胞會進一步導致神經元凋亡和BBB 損傷;應用AT1R 拮抗劑坎地沙坦(candesartan)則可抑制AngⅡ介導的神經炎癥[57-58]。Park 等[59]研究發現,長期輸注AngⅡ會通過小膠質細胞活化誘導抑郁樣行為。針對AngⅡ的促炎作用,有研究指出,降低小膠質細胞上AT1R/AT2R比例或激活ACE2/Ang(1-7)/MasR 軸,有助于抑制小膠質細胞激活,緩解神經炎癥[60-61]。
4 血管炎癥背景下BMEC誘導BRAS失衡
研究表明,血管病理損傷通常先于認知功能障礙和神經變性,是AD 的早期病理特征[62-63]。BRAS 作為重要的血管調控系統,其失衡是AD 早期血管病變的重要標志[5]。目前,誘導BRAS 失衡的具體機制尚不清晰,但可以肯定的是,其與AD 相關血管風險因素如高血壓、糖尿病、心力衰竭和動脈粥樣硬化等密切相關[15]。值得注意的是,在上述病理背景下,腦血管損傷程度較輕,BBB 仍保留著完整的結構和屏障功能,因此有毒有害物質難以入腦,但BMEC 長期暴露于循環系統中的炎癥因子的刺激下則會通過NVC 影響NVU 中的神經細胞,進而導致認知障礙。Tan等[64]研究發現,BMEC 分泌的信號素3G 可通過神經菌毛素2/叢狀蛋白A4 調控海馬突觸結構和可塑性。Lee 等[65]也發現,BMEC 可通過Notch 信號調控星形膠質細胞谷氨酸轉運體1的表達。這些發現進一步表明,BMEC 具有逆向調節上游神經細胞,影響NVU的作用。
在NVU 中,星形膠質細胞與BMEC 關系最為密切,星形膠質細胞通過其細胞終足與BMEC 緊密相連,共同構成BBB,并可向BMEC 傳遞神經遞質和信號分子,在神經元與BMEC 間發揮重要的橋梁作用[66]。因此,星形膠質細胞可能是BMEC 影響NVU 調控網絡的主要靶細胞。BMEC 受炎癥刺激時,會表達白細胞介素6(interleukin 6,IL-6)和IL-1β 等炎癥因子,而IL-6 和IL-1β 具有促進星形膠質細胞中AGT 表達和提高中樞AngⅡ水平的作用[67-68]。上述研究結果提示,在血管炎癥背景下,BMEC 可通過釋放炎癥因子,促進星形膠質細胞中AGT 的表達,干擾BRAS 穩態,最終導致NVU 失調(圖4)。
5 外周RAS通過BMEC調控BRAS
循環系統RAS 失調是外周血管性疾病(如高血壓)的主要特征之一,具體表現為ACE 活性提高和血漿AngⅡ濃度升高,而循環系統中較高濃度的AngⅡ具有促炎活性,會進一步誘導血管炎癥[69-70]。而BMEC 既是BRAS 重要的效應細胞,也是外周AngⅡ重要的靶細胞。因此,在高血壓背景下,BMEC 可在AngⅡ的持續刺激下,通過IL-6和IL-1β等炎癥因子及內皮素的表達與釋放,直接調控鄰近星形膠質細胞AGT 表達,影響BRAS 穩態[67-68],或通過抑制硫化氫及NO 的產生和釋放,間接影響BRAS的生理功能[69-74]。內皮素同樣具有縮血管活性,可與AngⅡ協同發揮更強、更持久的縮血管作用,而硫化氫和NO水平的降低則會抑制ACE2的表達,進一步促進中樞AngⅡ水平的升高[75]。AngⅡ水平升高一方面會導致神經炎癥,另一方面也會加劇BBB 的損傷,介導毒性物質入腦,致使惡性循環,最終導致神經退行性病變的發生[7]。由此可見,BMEC 可能是實現外周RAS 與BRAS 內外呼應的重要橋梁,關注BMEC 在BRAS 調控中的作用既有利于闡明NVU的調控機制,同時也有利于理解心血管風險因素對AD病理的影響。
6 結語
BRAS 是CNS 中的重要調節系統,主要通過免疫應答和氧化應激調控NVU中的細胞狀態和功能。臨床證據表明,BRAS 失衡是AD 患者重要的病理特征,病理狀態下的AngⅡ可通過AT1R 促進CNS中Aβ 的產生以及細胞氧化應激和炎癥反應,加劇AD 病理進程。而RAS 抑制劑類藥物,特別是AT1R阻斷劑對延緩AD 病程、改善認知障礙具有積極作用,有望成為AD 治療的新策略。此外,本文依據NVC的調節網絡,總結了外周炎癥背景下BMEC通過影響中樞星形膠質細胞中AGT表達,進而干擾BRAS 穩態和誘導NVU 失衡的可能機制,并提出外周RAS借助BMEC與BRAS內外串擾的機制假說。該假說為解釋外周疾病誘導神經損傷的病理現象、深入探究NVU 調控網絡及開發AD 防治新策略提供了理論依據,值得進一步研究驗證。
猜你喜歡
中成藥(2021年5期)2021-07-21 08:39:04
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
中成藥(2018年6期)2018-07-11 03:01:24
中成藥(2018年5期)2018-06-06 03:11:43
天然產物研究與開發(2016年6期)2016-06-05 10:29:26
西南軍醫(2016年6期)2016-01-23 02:21:19
新疆醫科大學學報(2015年10期)2015-12-26 12:33:30
吉林大學學報(醫學版)(2015年4期)2015-12-17 07:48:13
實用中西醫結合臨床(2015年7期)2015-02-28 16:30:23
癌變·畸變·突變(2015年3期)2015-02-27 06:15:09
