
電感耦合等離子體發射光譜法測定催化劑中釩、鉀含量
2023-12-06 13:11:58徐臣松羅昊楠田坤
山東化工 2023年20期
徐臣松,羅昊楠,田坤
(貴州威頓催化技術有限公司,貴州 銅仁 554300)
硫酸是一種重要的化工原料,在石油化工、金屬冶煉、軍工及化肥等領域有著廣泛的應用,生產硫酸的原料一般有硫磺、硫鐵礦和冶煉煙氣等經高溫灼燒產生SO2氣體,產生的氣體與O2在轉化塔催化劑床層上轉化為SO3[1-2],再經水合制成硫酸。硫酸生產所使用的催化劑是釩系催化劑,因其具有良好的機械強度、活性以及穩定性等被廣泛使用。釩系催化劑中的主要活性成分為V2O5,通常含量為5%~10%。可降低硫酸生產過程中所需的活化能,此外,加入少量的助催化劑堿金屬鹽,可提高活性物質的活性、選擇性以及穩定性,釩含量、釩鉀含量等是影響催化劑低溫活性的關鍵因素,因此對催化劑中相關物質含量檢測具有重要作用[3]。對催化劑的性能有較大益處。
目前,關于釩系催化劑中釩、鉀含量的檢測,主要采用硫酸亞鐵銨滴定法和四苯基硼酸鉀重量法進行測定[4-6],其涉及過程較為繁瑣、復雜,分析過程較長,不能在短時間內獲得樣品相關組分含量數據、勞動強度大。在實際分析過程中,為提高數據的置信度和可靠性,因而對作業人員的專業素養和能力要求也很高。ICP的工作原理與原子熒光和原子吸收相似,其核心都是待檢原子在高能環境條件下,原子吸收特定能量的粒子的光子,使原子核外處于不同能級的電子從低能量狀態(基態)或亞穩態發生躍遷形成激發態,從而發射出特征光譜或形成吸收光譜。通過檢測發射特征光譜的波長或頻率確定原子的種類及通過檢測特征光譜的強度確定其相對含量。
近年來,電感耦合等離子體發射光譜(ICP-OES)測試方法作為組分含量檢測的一種重要手段,廣泛用于催化劑生產及廢催化劑回收利用過程中各組分含量的檢測和確定[7-8]。就二氧化硫氧化制酸用釩催化劑及工業硫酸廢催化劑的檢測分析有部分報道,但尚未將電感耦合等離子體發射光譜完全、深入地用于催化劑設計研發、生產制造,主要是因為催化劑基體組分結構復雜、硅載體類催化劑消解難度大、元素間存在干擾等[9]。儀器分析采用的是參比法,具有較高的靈敏度和響應度、基體效應小、線性范圍寬。其分析過程處理制樣時間較長,檢測過程較為簡單,且能在短時間內獲得多項數據,可為企業產品質量控制和設計研發提供有力的支持。為此,針對釩系催化劑建立一種快速測定組分含量的方法,對催化劑的生產質量控制有著重要意義,也可為企業開源節流提供一種新思路。
1 實驗部分
1.1 儀器
ICAP PRO電感耦合等離子體發射光譜儀(美國賽默飛世爾公司),工作條件見表1所示。YT系列微波消解儀、石墨消解儀,山東云唐智能科技有限公司;電熱恒溫烘箱,滬南電爐烘箱廠;多功能電動單道移液器,Discovery-E+、上海力辰邦西儀器科技有限公司。
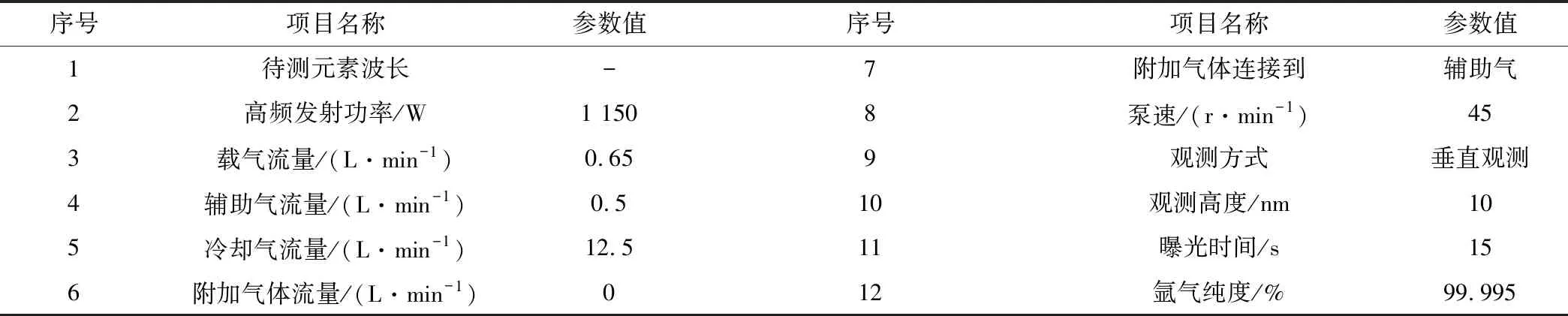
表1 ICP-OES儀器工作條件
1.2 試劑
硝酸、氫氟酸、鹽酸均為優級純;氫氧化鈉為優純級;實驗用水電阻率18.2 Ω由重慶力德高端水處理設備研發有限公司提供Limit-UF-20-LST型制備;釩單元素標準儲備液(1 000 mg/L)國家有色金屬及電子材料分析測試中心,鉀單元素標準儲備液(1 000 mg/L)北京北方偉業計量技術研究院。實驗中所用的100 mg/L的釩、鉀標準液均由1 000 mg/L釩、鉀標準儲備液稀釋制備,某廠提供催化劑2份。
1.3 實驗方法
1.3.1 器具材料準備
涉及樣品組分含量分析時,樣品的前處理是較為關鍵的一步,如果前處理失敗或引入其他雜質,對檢測結果有較大的影響。樣品前處理前首先要保證實驗室臺面清潔和干凈;其次是根據實驗需要準備實驗器材,本實驗需要測定催化劑樣品中的釩、鉀相關元素含量,以避免容器自身附帶目標元素或者干擾元素而造成分析結果偏離目標值。因此應選用聚四氟乙烯(PTFE)或氟塑料(PFA,帶有全氟化的烷基側鏈的四氟乙烯);隨后將所需使用到的實驗器具,用20%~30%的硝酸溶液浸泡一段時間,再用超純水漂洗干凈后,置于電熱恒溫干燥箱中烘干后使用。
1.3.2 樣品前處理
取適量已制備好的催化劑樣品,預先在105~110 ℃的恒溫干燥箱烘2 h并在干燥器中冷卻至室溫。
稱取0.1 g催化劑試樣置于消解罐內,加入5 mL硝酸、3 mL氫氟酸、2 mL鹽酸,將消解罐置于微波消解儀中,程序升溫為室溫到100 ℃,保持時間20 min,由100 ℃升至120 ℃,保持時間10 min,再由120 ℃升溫至180 ℃,保持時間20 min,待樣品完全溶解冷卻至室溫,若不能消解完全,則再加2 mL氫氟酸置于石墨消解儀加熱5 min,待溶解后進行趕酸處理。
試樣冷卻后轉移至250 mL容量瓶中,加水定容搖勻,此溶液為試驗母液A。
采用重量法稱量試驗溶液A,加入3%硝酸溶液,再稀釋5倍得到待測樣品。 同時做空白試驗。
2 結果與討論
2.1 分析譜線選擇
ICP-OES測試中,干擾類型有化學干擾、物理干擾、記憶效應、電離干擾及光譜干擾。在ICP測試過程中,光譜干擾存在于ICP中或者引入物質都會發射相當豐富的譜線,其中光譜干擾是最嚴重的干擾類型,分析譜線的選擇會直接影響測試結果,另外一種光譜干擾是由于基體成分及ICP光源本身所發射的強烈雜散光的影響而造成[10],對于此類背景干擾,可用ICAP PRO背景扣除,從而減小干擾。一般來說,分析譜線的選擇應滿足靈敏度高、背景低、干擾小的譜線作為分析譜線[11],此實驗過程采用鉀766.490 nm 、釩309.311 nm作為分析譜線。
2.2 酸體系選擇
ICP分析中的樣品一般是溶液狀態,因此選用何種消解酸體系將催化劑樣品轉化為測試溶液是ICP測試中一項十分重要的工作。酸的選擇需要根據試樣及待測元素性質選用,催化劑由硅藻土、堿金屬鹽、活性組分組成,硅藻土含有大量的SiO2·nH2O,使用硫酸消解可能有一部分目標元素存在于硅晶格中,由于硅晶格較穩定,只有使用氫氟酸才能破壞其晶格[12]。
試樣溶液經過蠕動泵注入到同心霧化器中,高速的氬氣將溶液吹散,形成均勻的氣溶膠進入霧化室,大液滴經霧化室壁經排液管進入到廢液收集桶,小液滴進入矩室進行分析。在此測試中過程中,酸體系的黏度、表面張力等對其精密度和準確度存在顯著影響。在使用高黏度酸時,其霧化效率低,進入等離子體的分析樣均勻性等受影響,導致測量值比真實值低,在相同酸度時,鹽酸、硝酸的黏度小,硝酸具有強氧化性,可將試樣中的有機和無機物質中的元素轉化為離子狀態,便于后續分析。因此,該實驗用酸體系:鹽酸+硝酸+氫氟酸。
2.3 消解方法的選擇
對某廠催化劑樣品,將樣品按照測定要求混合均勻,用四分法分取約40 g,在瓷研缽內破碎研細,再用四分法分取約20 g,繼續研磨,使全部通過150 μm(100目)試驗篩(符合GB/T 6003.3—1997中R40/3系列)。采用3種方法進行消解處理,結果見表2所示。

表2 三種消解方法的比較
取適量已制備好的催化劑樣品,預先在105~110 ℃的恒溫干燥箱烘2 h并在干燥器中冷卻至室溫。
稱取催化劑試樣0.2 g;記為m(精確至0.000 1 g)于預先稱有0.5 g氫氧化鈉的鎳坩鍋中,再稱取2.5 g的氫氧化鈉覆蓋試樣,加少許乙醇沖洗坩堝內壁濕潤。消解方法一:斜放上坩堝蓋(保證留有一定縫隙)置于馬弗爐中,逐級升溫至600~650 ℃,完全熔融后取出冷卻。
消解方法二:斜放坩堝蓋(保證留有一定縫隙)置于電爐上:開最小火,蒸發無水乙醇至坩堝中心氫氧化鈉焦黑;轉中火,直至坩堝中氫氧化鈉冒大氣泡;轉大火,直至坩堝中氫氧化鈉冒細小氣泡;調到最大火,直至坩堝中氫氧化鈉完全熔融呈液態,保持10 min,取出冷缺。用熱水沖洗坩堝外壁,置于盛有100 mL沸水的四氟燒杯中,待試樣溶解后,加入10 mL硝酸,待試樣完全溶解后用熱水洗出坩堝,并沖洗燒杯內壁,燒杯放入水中稍冷。
消解方法三:稱取0.1 g催化劑試樣置于消解罐內,加入5 mL硝酸、3 mL氫氟酸、2 mL鹽酸,將消解罐置于微波消解儀中,程序升溫為室溫到100 ℃,保持時間20 min,由100 ℃升至120 ℃,保持時間10 min,再由120 ℃升溫至180 ℃,保持時間20 min,待樣品完全溶解冷卻至室溫,若不能消解完全,則再加2 mL氫氟酸置于石墨消解儀加熱5 min,待溶解后進行趕酸處理。試樣冷卻后轉移至250 mL容量瓶中,加水定容搖勻,此溶液為試驗母液A。
采用重量法稱量試驗溶液A,加入3%硝酸溶液,再稀釋5倍得到待測樣品。 同時做空白試驗。
吸出樣時,霧化器產生的部分氣溶膠會通過噴槍組件傳遞到等離子體,氣溶膠在等離子體上產生負載,引起等離子體條件變化,使樣品分解成原子或者離子,經電子射頻(RF)產生的等離子體激發,當原子和離子由高能態躍遷為低能態時,每一種元素都會發出特定的波長,儀器通過檢測光強度確定元素含量。該過程涉及的重要組件中心矩管直徑2 mm,當樣品溶液中存在沉淀時,會造成中心矩管及進樣系統堵塞,從而造成檢測靈敏度、精密度及準確度下降。故該實驗采用消解方法三進行試驗。
2.4 標準曲線的配置
分別移取0,0.5,1,2,4,8 mL濃度為100 mg/L的釩、鉀單元素標準液于6個50 mL容量瓶中,用體積分數3%硝酸溶液定容至刻度、搖勻,配置成0,1.0,2.0,4.0,8.0,16.0 mg/L的混合標準液,在儀器工作條件下構建相應待測元素的工作曲線,測試10次樣品空白,計算其標準偏差,3倍的標準偏差即為方法的檢出限,本方法測定的曲線方程及檢出限見表3所示。

表3 曲線方程、相關系數及檢出限
由表3中的測試數據可知,釩、鉀待測元素的相關系數為0.999 9以上,為測試結果數據提供了置信度和可靠性的基礎,檢出限分別為0.001,0.022 1 μg/mL,采用的標曲溶液均在檢出限以上,可滿足樣品的測試需求。
2.5 精密度測試
采用實驗方法,實驗室通過選擇與測試樣品相似的質控樣,能夠有效地監控和保障結果的準確度,尤其是在確保前處理效果,驗證回收率,降低基質干擾等方面。對質控樣重復測定10次,結果見表4所示。

表4 方法精密度測試(n=10)
由表4檢測數據可知,采用鉀、釩標準物質按新方法進行測定監控,連續測10次,根據標準物質提供的可靠的量值,不確定度及穩定性和均勻性,對測定結果進行校正及其背景扣除,其相對標準偏差(RSD)分別為0.3%,0.7%,催化劑中待測元素相對標準偏差(RSD)均小于1.0% ,分析精密度方法可滿足測試的要求。
2.6 準確度測試
利用未知樣和標準物質進行加標回收實驗照,測定目標元素的發射強度,根據標準曲線確定目標元素的含量,利用目標元素的加入值與實測值進行比較來確定方法的準確度,其結果見下表5所示。
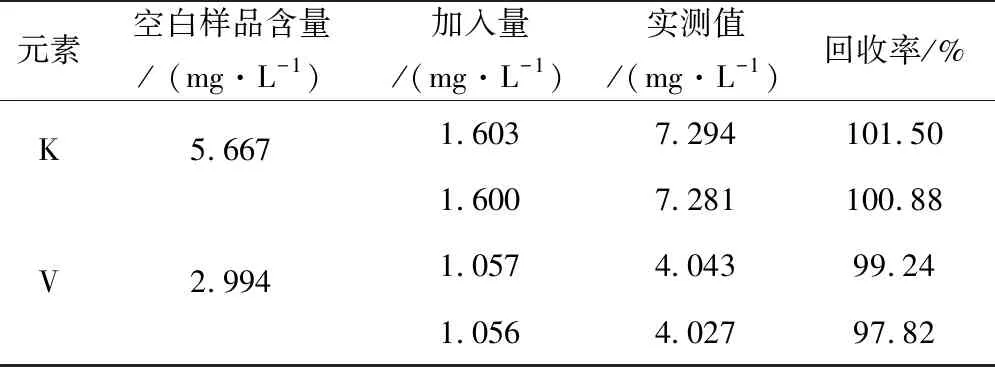
表5 催化劑加標回收率
由表可知,催化劑中待測元素K、V加標回收率在97.82%~101.50%間,表明使用該方法對催化劑成品中的K、V元素的檢測準確度、精密度滿足測試的要求。
2.7 樣品測定
催化劑中待測元素含量測定分別采用硫酸亞鐵銨標準溶液滴定化學分析法、四苯硼鉀法分析法測定值與ICP測定未知樣的分析結果對比見表6所示。

表6 催化劑組成測定結果
由表6可知,采用ICP-OES法測定催化劑元素含量,V、K目標元素的測定與化學測定法所得的結果相近。二氧化硫氧化制酸用釩催化劑生產涉及到多步控制,生產過程組分含量對催化劑產品的質量具有重要的影響,因此快速檢測和監測涉及過程樣品含量數據,為生產控制及工藝調整提供數據指導,能保障生產過程控制的及時性,提高工藝控制效率。目前,催化劑中釩、鉀含量采用硫酸亞鐵銨滴定法和四苯硼鉀法測定,所需時間5.5 h,而采用ICP法測定釩、鉀含量所需時間4 h(制樣,分析過程),兩法相比,采用ICP法測定時間節省27.27%,極大縮短樣品分析時間,降低成本,操作步驟簡單,易于操作。因此ICP應用于企業生產管理可提高生產效率,規范生產行為,節能降耗等,為企業產品質量提供重要保障,讓企業在市場競爭中處于有利地位。
3 結論
實驗結果表明:采用ICP-OES法測定催化劑中釩、鉀含量的實驗中,方法操作較簡單,干擾少, 流程短,相對標準偏差(RSD)值小于1.0%,加標回收率在97.82%~101.50%間,具有較好的精密度和準確度,能夠滿足催化劑中釩、鉀含量的測定要求。在傳統化學檢測的基礎上,采用電感耦合等離子體發射光譜(ICP-OES)的測試方法,能一次獲得多個檢測對象的含量指標,能在較大程度上降低工作量和勞動強度,節省檢測時間。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:45:06
