聯吡啶二酰胺配體對U(VI)/Mo(VI)的萃取分離及機理
2024-01-08 04:01:40修濤元張萌葉國安矯彩山袁立永石偉群
哈爾濱工程大學學報 2023年12期
關鍵詞:分配
修濤元, 張萌, 葉國安,3, 矯彩山, 袁立永, 石偉群
(1.哈爾濱工程大學 核科學與技術學院,黑龍江 哈爾濱 150001; 2.中國科學院 高能物理研究所, 北京 100049; 3.中國原子能科學研究院, 北京 102413)
醫用放射性同位素在疾病的診斷和治療中發揮著非常重要的作用,其中99mTc藥物被廣泛應用于疾病診斷[1-3]。99mTc主要由其母核素99Mo通過99Mo/99mTc發生器制備[4-5]。目前,99Mo主要獲取方式有反應堆生產、加速器制備和中子發生器制備等[6-7]。其中利用反應堆中高濃縮鈾/低濃縮鈾靶體的裂變產生99Mo是最主要的方式[8-9]。制備后靶件溶解液中含有大量鈾和裂片元素,其中U(VI)和Mo(VI)化學性質相似,這使得U(VI)/Mo(VI)分離成為一個難題。因此,研究U(VI)/Mo(VI)分離具有科學價值和現實重要意義。
U(VI)/Mo(VI)的分離方法有萃取法、離子交換法、沉淀法、吸附法和揮發法等[10]。溶劑萃取因其條件溫和、操作簡單、易于產業化而廣泛應用于核燃料循環前后端[11-12]。Lsasheen等[13]將LIX 622N用于從含有Mo(VI)和U(VI)的硫酸鹽浸出液中萃取回收Mo(VI),并獲得高純度的MoO3(99.99%)。Behera等[14]用Alamine 310、TBP、DPSO及其苯中的混合物從磷酸水溶液對鈾鉬分離進行研究,Alamine 310、TBP和DPSO可以實現分離,其中DPSO在8 mol/L H3PO4下,U(VI)/Mo(VI)分離因子最大可達到180.37。Behera等[15]用有機磷酸、次膦酸及其硫代衍生物在 0.1~1.0 mol/L 的HCl下萃取分離U(VI)/Mo(VI),比較了 PC-88A、Cyanex 272、Cyanex 301和Cyanex 302的萃取分離U(VI)/Mo(VI)效果,Cyanex 301在1 mol/L的HCl 下可以實現U(VI)/Mo(VI)分離。許多配體設計出,并用于分離U(VI)/Mo(VI),但大多數配體含有磷,不符合二次廢物最小化的“CHON”原則[16]。氮雜環酰胺類配體用于萃取分離錒系元素。其中具有代表性的有吡啶二酰胺類、聯吡啶二酰胺類和鄰菲啰啉二酰胺類等。該類配體在酸性和輻照條件具有穩定性好、動力學快和合成簡單等特點。同時,這類配體又符合“CHON”原則,因此得到廣泛關注和研究。其中聯吡啶二酰胺類配體自身柔性較大,可以通過自身的旋轉更好地與金屬離子配位。目前,聯吡啶二酰胺配體在錒系元素分離中的潛力越來越受到人們的關注。該類配體沒有相關文獻進行U(VI)/Mo(VI)分離研究。
本文采用3種不同烷基取代基的聯吡啶二酰胺配體(Et-EB-DABP、But-EB-DABP和Oct-EB-DABP)作為萃取劑。利用溶劑萃取法,在不同稀釋劑、酸度、時間、配體濃度和U(VI)濃度等條件下,對U(VI)/Mo(VI)分離進行系統研究;選用H2O、HNO3(0.01 mol/L)和Na2CO3(5%) 3種反萃劑進行反萃實驗;通過傅里葉變換紅外光譜(FT-IR)和電噴霧電離質譜(ESI-MS)進行配位機理研究。
1 配體合成及實驗方法
1.1 配體合成
配體合成路線如圖1所示。將化合物1 (4.69 g,0.025 mol)和KMnO4(26.2 g,0.17 mol)加入到300 mL水中;將混合物加熱至90 ℃攪拌14 h;冷卻至室溫后過濾,將濾液用Et2O萃取3次;所得水溶液通過1 mol/L的HCl調至pH為2,過濾干燥后的得到白色化合物2[17]。將SOCl2(25 mL)加入裝有化合物2 (1.00 g,4.10 mmol)的燒瓶里,85 ℃回流4 h;通過旋蒸除去多余的SOCl2,得到黃色化合物3[18];化合物3溶于CH2Cl2(25 ml)中,將NEt3(1.66 g,16.40 mmol)加入到CH2Cl2(10 ml)并分別加入化合物4 (1.74 g, 9.02 mmol)、5 (2.00 g, 9.02 mmol)或6 (2.50 g, 9.02 mmol)[19],化合物4、5和6合成方法參照文獻[20],在冷CH4CH2OH浴下緩慢加入到化合物3的CH2Cl2溶液中;在40 ℃下回流4 h后,通過旋蒸除去溶劑,用硅膠柱層析法純化(展開劑: CH3OH/CH2Cl2=1/20),得到3種白色聯吡啶二酰胺配體:化合物7 (Et-EB-DABP(L1))、化合物8 (But-EB-DABP(L2))和化合物9 (Oct-EB-DABP(L3))。3種配體已經NMR (1H,13C) 和ESI-MS確認,如圖2所示。
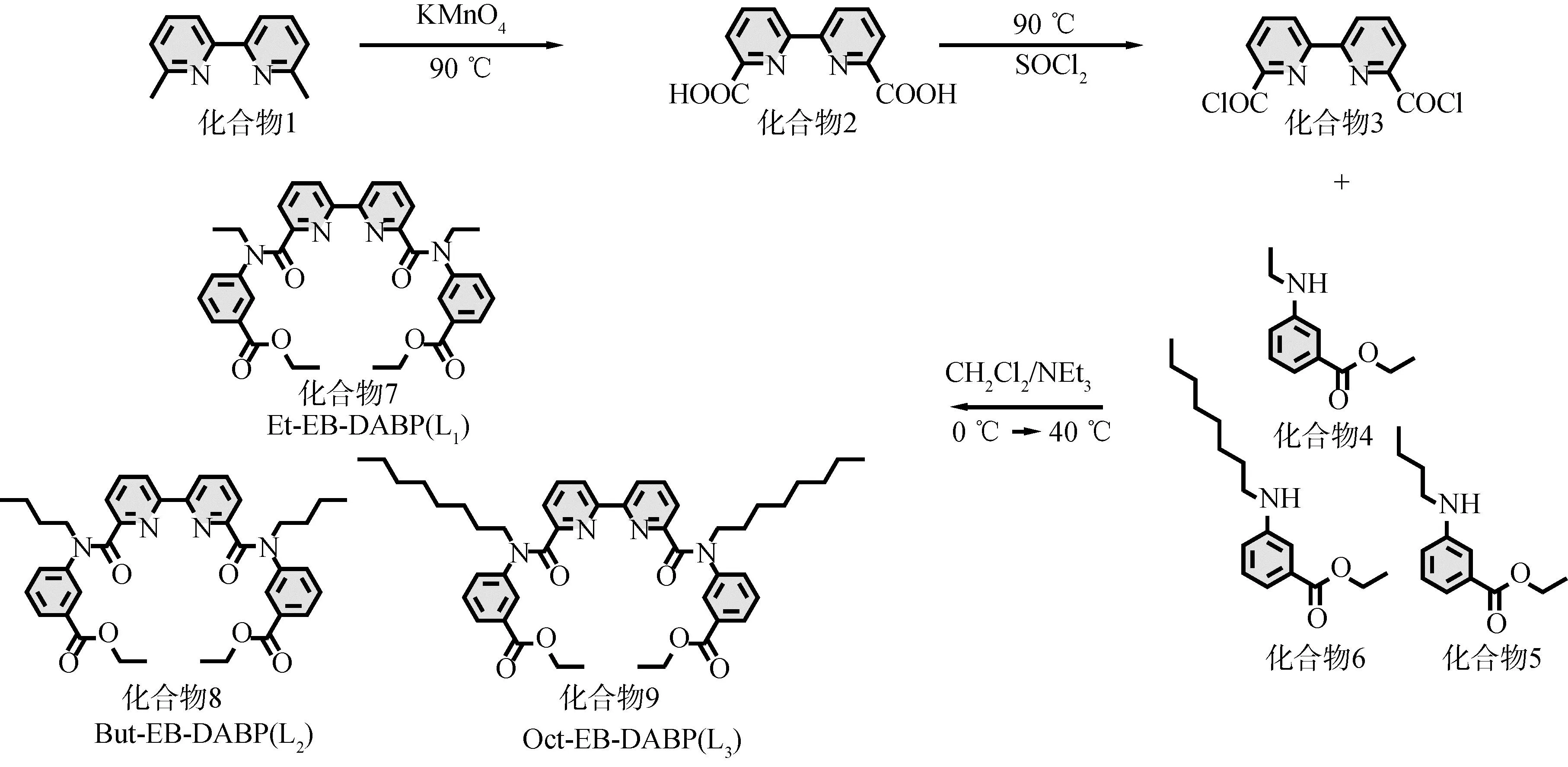
圖1 配體合成路線Fig.1 The synthetic route of the ligands

圖2 3種配體(L1, L2和L3)的NMR(1H, 13C)和ESI-MS譜圖Fig.2 The NMR(1H, 13C) and ESI-MS spectra of three ligands (L1, L2, and L3)
1.2 萃取及光譜實驗方法
將一定量UO2(NO3)2·6H2O和Na2MoO4分別溶于1 mol/L的HNO3并用容量瓶定容,制備U(VI)和Mo(VI)母液。根據不同實驗條件配置不同水相。將一定量的L1、L2和L3分別溶于不同稀釋劑得到有機相。預平衡有機相,將有機相與水相混合,在室溫下充分振蕩30 min, 相比(O/A)始終為1,進行萃取和反萃。經7 000 r/min離心分離3 min后,取水相,用電感耦合等離子體發射光譜儀(ICP-OES)測定U(VI)和Mo(VI)的濃度。有機相U(VI)和Mo(VI)的濃度通過差減法得到。分配比D和分離因子β為[21-22]:
D=[M]org/[M]aq
(1)
βU/Mo=DU/DMo
(2)
式中:[M]org和[M]aq分別表示萃取后有機相和水相中的金屬離子濃度。
反萃實驗選用Na2CO3(5%)、H2O和HNO3(0.01 mol/L)作為反萃劑。將萃取后有機相與反萃劑等體積混合,離心后測定水相中金屬離子的濃度。反萃率S為[20]:

(3)
式中:[M]str.aq和[M]ext.aq分別為反萃后水相中的金屬離子濃度和反萃前有機相中的金屬離子濃度。
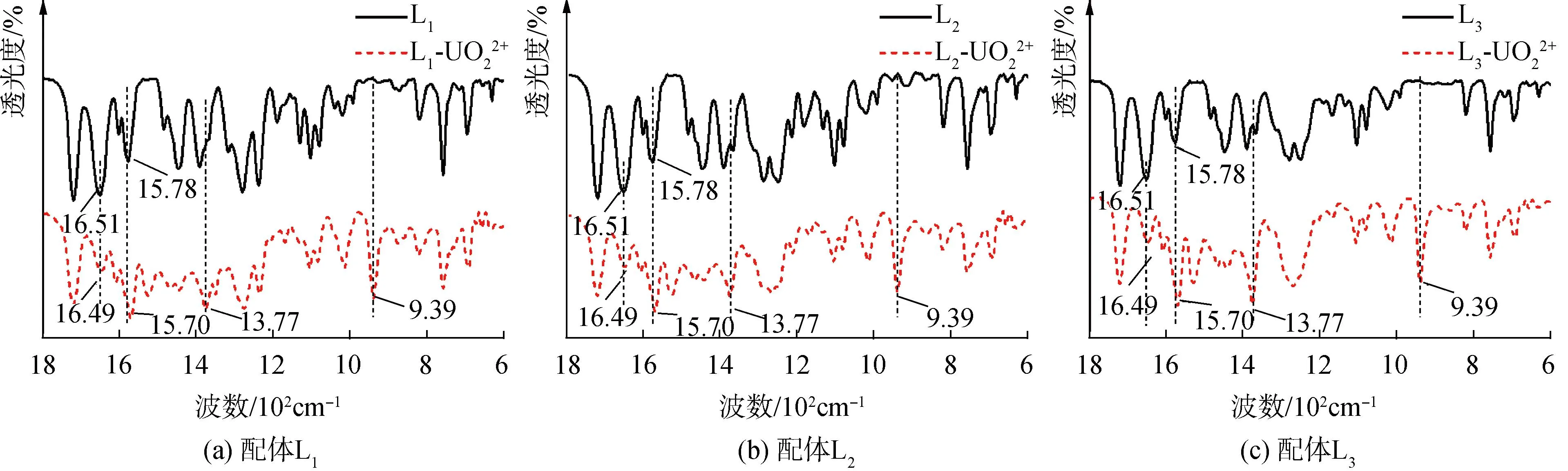
萃取前后FT-IR通過的將L1、L2和L3萃取U(VI)前后的有機相采用溴化鉀壓片法在500~2 000 cm-1波長進測試。取一定量的L1、L2和L3分別溶于乙腈中配置濃度為0.01 mmol/L的母液。取1 mL配體乙腈溶液并逐次加入2.5 μL的1 mmol/L U(VI)乙腈溶液調節U(VI)與配體比例。將上述溶液置于恒溫振蕩器中震蕩30 min,以確保配位平衡。隨后,對溶液進行ESI-MS測試。
2 萃取結果與機理分析
2.1 稀釋劑的影響
為了研究配體在不同溶劑中的萃取性能,選用5種不同稀釋劑(正辛醇、甲苯、二氯甲烷、1,2-二氯乙烷和間硝基三氟甲苯)進行萃取實驗。萃取條件為:[L1]=[L2]=[L3]=2 mmol/L、[HNO3]=4.00 mol/L、[U(VI)]=0.5 mmol/L、t=30 min、T=298 K和O/A=1。不同稀釋劑的影響圖3所示,3種配體對U(VI)具有較好的萃取效果,其分配比由低到高依次為:正辛醇<甲苯<二氯甲烷<1,2-二氯乙烷<間硝基三氟甲苯。由于間硝基三氟甲苯對聯吡啶二酰胺類配體具有良好的溶解度,同時具有良好的疏水性和較高介電常數,可提高萃取能力[20]。因此,選擇間硝基三氟甲苯作為后續實驗的稀釋劑。

圖3 稀釋劑對3種配體(L1, L2和L3)萃取U(VI)和Mo(VI)的影響Fig.3 Influence of diluents on the DU value by L1, L2 and L3 ligands
2.2 HNO3濃度的影響
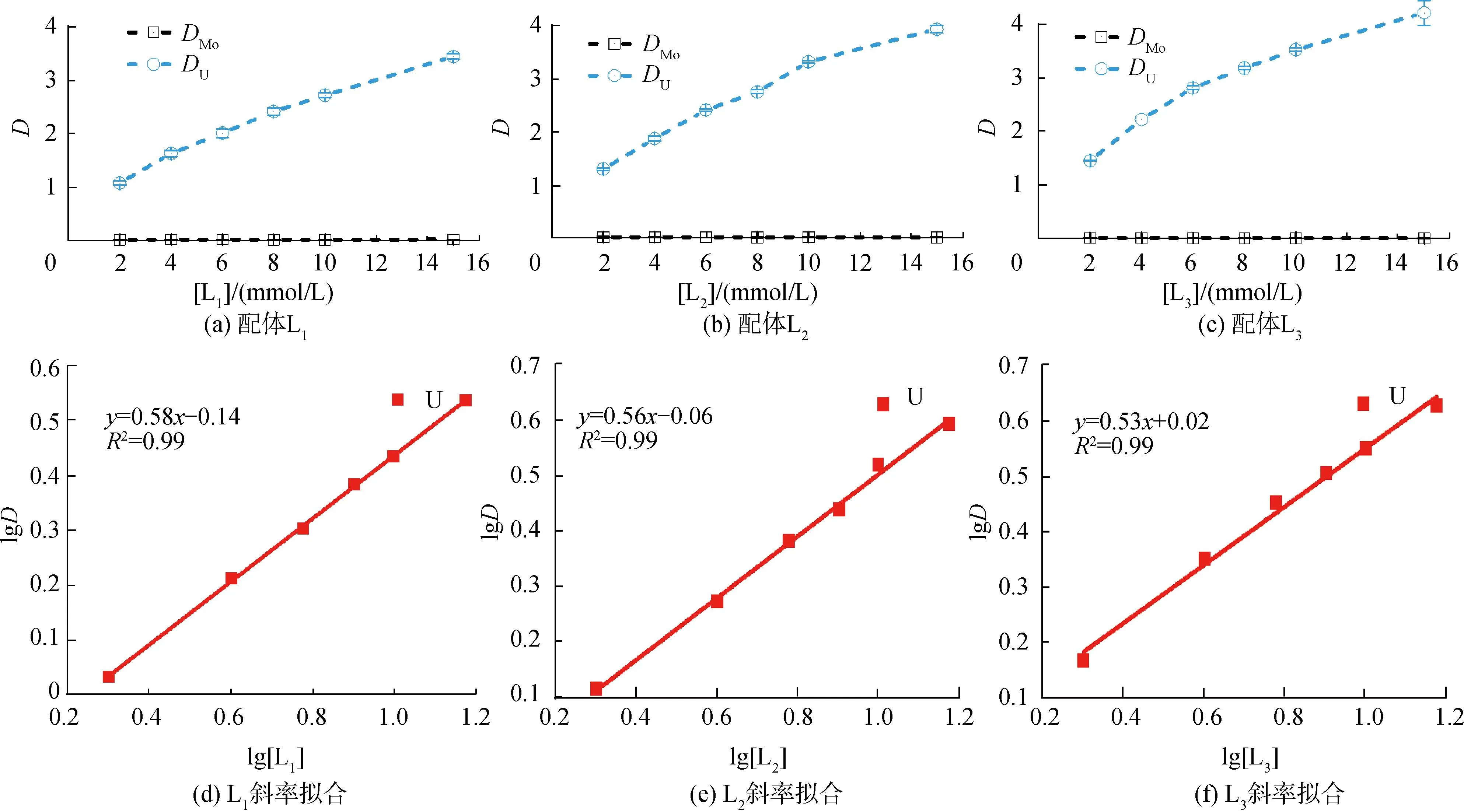
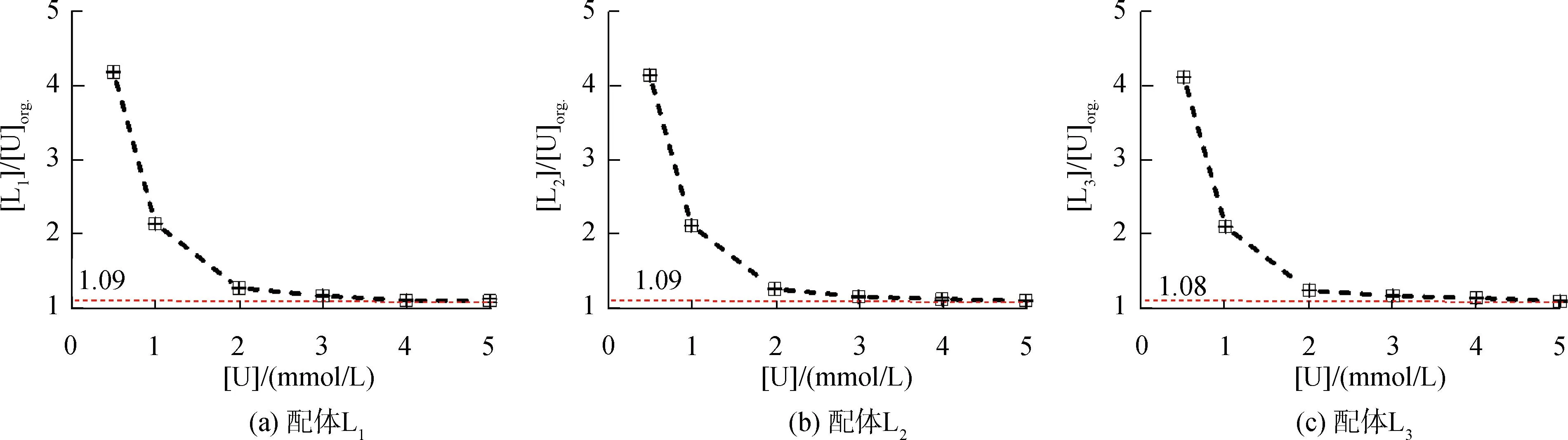
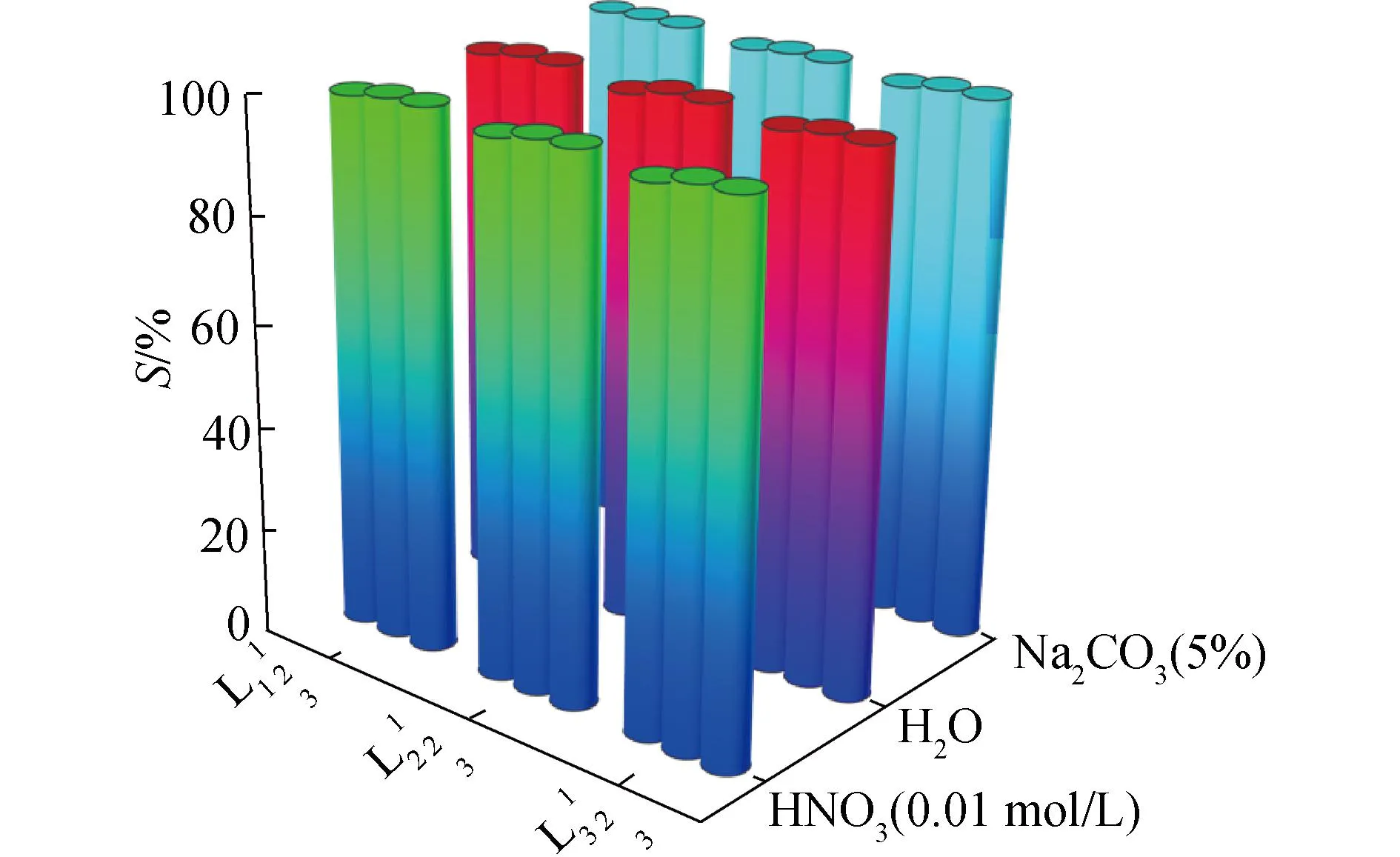
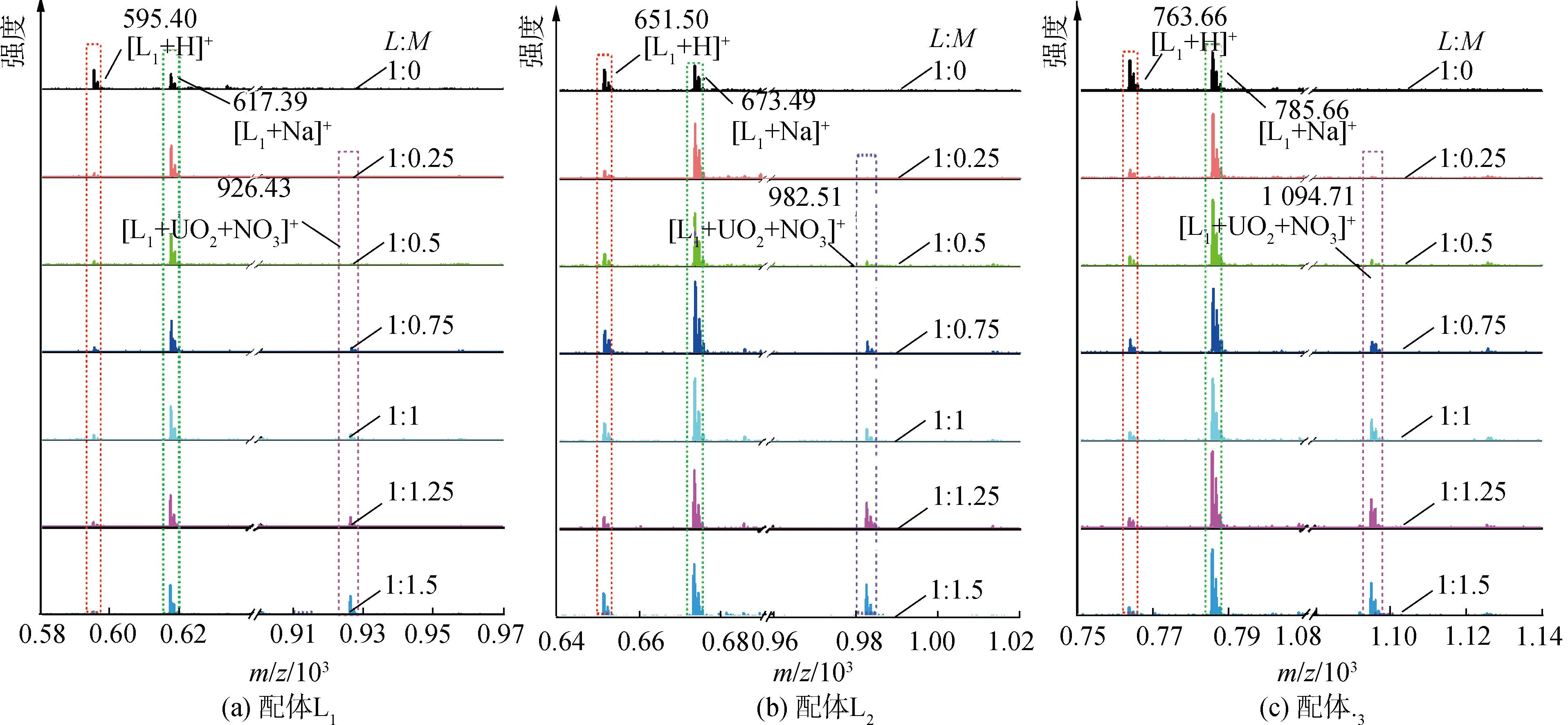
研究HNO3濃度的影響時,萃取條件為:[L1]=[L2]=[L3]=2 mmol/L、0.1 mol/L≤[HNO3]≤4 mol/L、[U(VI)]=0.5 mmol/L、[Mo(VI)]=0.2 mmol/L、t=30 min、T=298 K和O/A=1。DU、DMo和βU/Mo隨HNO3濃度變化如圖4所示。3種配體對U(VI)和Mo(VI)具有較強的分離能力,分離能力由大到小依次為L1 圖4 HNO3濃度對3種配體(L1, L2和L3)萃取U(VI)和Mo(VI)的影響Fig.4 Influence of HNO3 concentration on the D value and β value of U(VI) and Mo(VI) by L1, L2 and L3 ligands 萃取動力學是評價萃取性能的重要參數之一。研究接觸時間的影響時,萃取條件為:[L1]=[L2]=[L3]=2 mmol/L、[HNO3]=0.5 mol/L、[U(VI)]=0.5 mmol/L、[Mo(VI)]=0.2 mmol/L、1 min≤t≤30 min、T=298 K和O/A=1。接觸時間對U(VI)和Mo(VI)萃取的影響如圖5所示。隨著接觸時間的增加,Mo(VI)的分配比一直趨于零。L1、L2和L3對U(VI)的萃取在15 min內達到平衡。3種配體的U(VI)萃取動力學速度較快。與鄰菲啰啉酰胺類配體對比,動力學相對較慢,原因為聯吡啶骨架在萃取過程中,需要扭曲自身,從而需要更長的時間才能達到平衡[20]。 圖5 時間對3種配體(L1, L2和L3)萃取U(VI)和Mo(VI)的影響Fig.5 Influence of contact time on the D value of U(VI) and Mo(VI) by L1, L2 and L3 ligands 研究配體濃度的影響時,萃取條件為:2 mmol/L≤[L1]=[L2]=[L3]≤10 mmol/L、[HNO3]=0.5 mol/L、[U(VI)]=0.5 mmol/L、[Mo(VI)]=0.2 mmol/L、t=30 min、T=298 K和O/A=1。如圖6所示,隨著配體濃度的增加,配體對U(VI)的分配比迅速增加,而對Mo(VI)的分配比一直趨近于零。斜率分析法通常用于確定配體與金屬離子的化學計量比。由于L1、L2和L3對Mo(VI)的萃取效果較差,斜率分析法無法分析配體與Mo(VI)的化學計量比。因此,只研究了配體與U(VI)的化學計量比。在溶液中,萃取平衡等式為[22]: 圖6 不同濃度萃取U(VI)和Mo(VI)的影響Fig.6 Influence of ligand concentration on the D value of U(VI) and Mo(VI) 萃取平衡常數KexU和U(VI)的萃取分配DU為: 將式(4)~(6)重新組合得到: > (4) (5) (6) (7) 以分配比的對數值作為縱坐標,以萃取劑濃度的對數值作為橫坐標,可得到線性等式的斜率值為金屬離子與萃取劑分子形成配合物的化學計量比。如圖6所示,L1、L2和L3與U(VI)的斜率分別為0.58、0.56和0.53,說明在間硝基三氟甲苯溶劑中,L1、L2和L3與U(VI)在萃取過程中的化學計量比為1∶1,形成的配合物為:UO2(NO3)2·L。由于L1、L2和L3為四齒配體,易于占據鈾酰平面六配位中的4個位點,而其自身垂直方向存在2個氧原子阻止配體進一步配位,因此不能形成配體與鈾酰比例為2∶1配合物。 配體對金屬離子的萃取容量是衡量其萃取性能的重要指標。為了探究3種配體(L1、L2和L3)對U(VI)的萃取容量。將配體濃度設為2 mmol/L,通過改變U(VI)濃度探究其萃取容量。具體萃取條件為:[L1]=[L2]=[L3]=2 mmol/L、[HNO3]=4.0 mol/L、0.5 mmol/L≤[U(VI)]≤5.0 mmol/L、t=30 min、T=298 K和O/A=1。由圖7可知,當U(VI)濃度為5 mmol/L時,有機相中配體與U(VI)的比值接近于1。這表明3種配體對鈾的萃取容量均達到理論值[23]。 圖7 U(VI)濃度的影響Fig.7 Influence of U(VI) concentration 對于一種具有應用前景的配體,不僅要有良好的萃取性能,同時也有良好的反萃性能。選用HNO3(0.01 mol/L)、H2O和Na2CO3(5%)作為反萃劑。U(VI)的3級反萃如圖8所示。3種反萃劑的對U(VI)的反萃效果由高到低:Na2CO3(5%)>H2O>HNO3(0.01 mol/L)。用5% Na2CO3溶液,只需一步就可以達到接近100%的反萃率。這是由于與碳酸鹽相比,配體和U(VI)配合物的logβ相對較低[22]。因此,有機相中負載的U(VI)很容易被碳酸鹽剝離,從而形成更穩定的UO2(CO3)n[22]。3種反萃的3次反萃率均接近100%,表明配體具有良好的反萃性能。 圖8 3種不同反萃劑H2O、HNO3 (0.01 mol/L)和Na2CO3 (5%)對U(VI)的反萃Fig.8 Stripping of U(VI) from organic phase by ultrapure water, 0.01 mol/L HNO3 and 5% Na2CO3 圖9 L1、L2和L3萃取U(VI)前后的FT-IR譜圖Fig.9 The FT-IR spectra of ligands (L1, L2 and L3) and its complex with U(VI) 由圖10可知,L1、L2和L3這3種配體與H+結合的峰分別出現質荷比m/z為595.40、651.50和763.66,以及與Na+結合的峰,出現在m/z為617.39、673.49和785.66。隨著U(VI)的不斷滴加,3種配體的質譜譜圖中逐漸出現配體結合一個鈾酰一個硝酸根的配合物[L1+UO2+NO3]+(m/z926.43),[L2+UO2+NO3]+(m/z982.51)和[L3+UO2+NO3]+(m/z1 094.71),且U(VI)的比例越高,相應的峰越強。以上分析可知,U(VI)與L1、L2和L3這3種配體形成U(VI)∶L=1∶1的配合物。本文方法與斜率法結果相吻合。 圖10 CH3CN體系U(VI)滴定L1、L2和L3的ESI-MS分析Fig.10 ESI-MS titration of ligands (L1, L2 and L3) with U(VI) in CH3CN 1)3種聯吡啶二酰胺配體在最優條件下分離因子達到~2 000,實現U(VI)和Mo(VI)之間的高效分離。 2)研究發現配體烷基鏈越長,在稀釋劑中的溶解性越高,因此對U(VI)的萃取能力越強,進而表現出更強U(VI)/Mo(VI)的分離能力。 3)3種配體萃取動力學快,15 min內達到平衡。3種配體對U(VI)的萃取容量甚至達到理論值。3種反萃劑3次反萃百分比都接近100%,表明該類配體具有良好的循環利用潛力。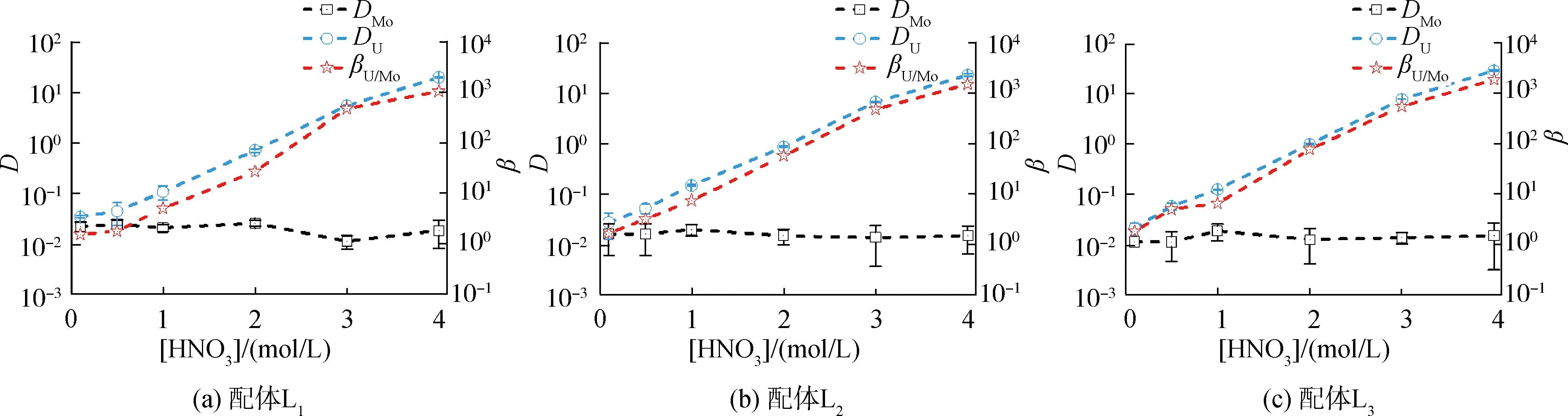
2.3 時間的影響

2.4 配體濃度的影響
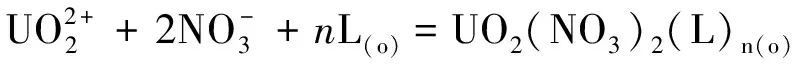
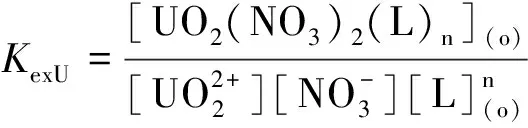
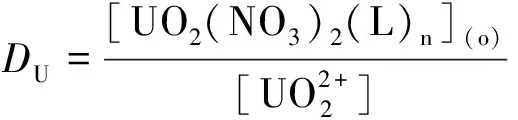
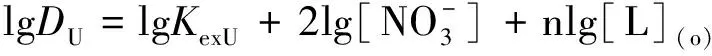
2.5 U(VI)濃度的影響
2.6 反萃
2.7 FT-IR分析

2.8 ESI-MS滴定分析
3 結論
猜你喜歡
天水行政學院學報(2022年4期)2022-11-18 09:02:36
艦船科學技術(2022年13期)2022-08-11 09:30:02
鐵道通信信號(2020年9期)2020-02-06 09:15:22
漢語世界(The World of Chinese)(2019年3期)2019-07-01 02:37:48
數學大王·趣味邏輯(2019年5期)2019-06-13 20:27:43
小學科學(學生版)(2019年5期)2019-05-21 01:00:18
中學生數理化·中考版(2018年10期)2018-12-07 00:44:52
經濟技術協作信息(2018年30期)2018-11-22 06:20:24
中央社會主義學院學報(2017年1期)2017-04-16 05:34:07
中國衛生(2014年12期)2014-11-12 13:12:40
