持續高糖狀態對Kv11.1離子通道蛋白表達的影響
2024-01-11 09:36:46韓穩琦陳海潮尤紅俊鄧紀釗
陜西醫學雜志 2024年1期
韓穩琦,王 毅,陳海潮,尤紅俊,鄧紀釗,祁 杰
(陜西省人民醫院心血管內二科,陜西 西安 710068)
長QT綜合征(Long Q-T Syndromes,LQTS)可由快速延遲整流鉀電流(Rapid component of delayed rectifier potassium current,IKr)的降低引起,IKr是人類ether-á-go-go相關基因(the human Ether-à-go-go-Related Gene,hERG)編碼的心臟快速延遲整流鉀通道(Kv11.1)α亞單位[1-4]。Ikr是動作電位復極期重要的外向離子流,各種因素及藥物致Kv11.1表達減少及通道異常使IKr振幅減低導致QT間期異常易引起心肌心內外膜和M層細胞復極不均一,從而發生尖端扭轉性室速(Torsade de pointes,TdP),甚至猝死[5-8]。研究發現糖尿病患者心臟中hERG通道的表達顯著下調,這種下調是糖尿病復極減慢和QT間期延長的關鍵因素[9-10]。然而,對糖尿病誘導的IKr/hERG功能障礙的認識尚不完全,然而,目前尚不清楚不同葡萄糖濃度hERG的表達及蛋白轉運是否發生改變。
本研究構建了以EGFP為報告基因的增強子真核表達重組質粒,脂質體轉染構建HEK239T模型,以期通過EGFP 的自發熒光特點對HERG蛋白表達進行實時定量檢測。本研究通過脂質體轉染EGFP標記得HERG基因在HEK239T模型中明確地證明pEGFP-N1-HERG在不同濃度葡萄糖干預后KV11.1的表達,為糖尿病患者長期高糖狀態時KV11.1通道異常導致QT間期延長的分子學機制提供科學依據。
1 材料和方法
1.1 實驗材料 pcDNA3-HERG-C2質粒(氨芐青霉素抗性)由Wisconsin大學BD Anson教授贈予,pEGFP-N1質粒(卡那霉素抗性)購自Sigma公司,EcoRI和HindⅢ限制性內切酶、BamHⅠ限制性內切酶及500~15000 bp DNA分子量標準ladder購自Fermentas公司,T4-DNA Ligase購自Invitrogen公司,DNA轉染試劑、質粒提取試劑盒均為Roche公司產品,DMEM培養基及胎牛血清為西安沃爾森公司產品,核酸染料和DH5α大腸桿菌購自西安昕泰生物公司,卡那霉素購自西安沃爾森生物公司。2000型凝膠成像系統:美國Biotech公司,HEK293T細胞株取自我校心臟離子通道病教育部重點實驗室,葡萄糖粉購自sigma公司,流式細胞儀(FALS CALIBAR BD公司),常用試劑為本實驗室配制。
1.2 實驗方法
1.2.1 真核表達載體pEGFP-N1-HERG的構建和擴增:利用HERG在GenBank基因序列基因兩側單一的EcoRI和HindⅢ限制性內切酶雙酶切pcDNA3-HERG及pEGFP-N1,同時進行瓊脂糖凝膠電泳驗證條帶位置,T4 DNA連接酶將含有HERG的片段和pEGFP-N1片段連接起來,重組成pEGFP-N1-HERG綠色熒光真核表達載體并進行堿基序列測序。新構建質粒采用大腸桿菌DH5α進行轉化,LB-卡那霉素培養基篩選新構建產物,37 ℃培養溫箱孵育20 h后挑選單菌落于25 ml LB-卡那霉素培養液中進行擴增,細胞裂解法提取質粒后再次將新構建真核表達載體pEGFP-N1-HERG送上海生工生物有限公司進行堿基序列測序驗證。
1.2.2 HEK293T細胞(Human embryo kidney cell)培養及質粒轉染:將HEK293T細胞置于37 ℃、5%CO2條件培養箱中用含10%胎牛血清的DMEM低糖培養基中培養。轉染前均勻種植于六孔培養皿中,24 h后待細胞貼壁生長至50%~70%時,采用脂質體轉染法。預混物包括無血清DMEM 100 μl、轉染劑4 μl,pEGFP-N1-HERG 3 μg。
1.2.3 葡萄糖干預及融合蛋白表達鑒定:質粒轉染4 h后給予不同濃度葡萄糖(5、17.5、30 mmol/L)干預細胞,24 h后換液再次給予以上濃度葡萄糖序貫干預細胞,48 h后PBS收獲細胞行流式細胞術檢測轉染效率及熒光強度參數進行分析HERG綠色熒光融合蛋白表達。每個樣品的質粒表達效率鑒定以流式細胞儀在488 nm波長激發下,于530 nm處獲取1×104個細胞的平均綠色熒光值進行分析。

2 結 果
2.1 綠色熒光表達載體pEGFP-N1-HERG的構建 用HindⅢ和EcoRI限制性內切酶雙酶切pcDNA3-HERG和pEGFP-N1。瓊脂糖凝膠電泳定位,pEGFP-N1為一條目的條帶,約4000 bp和5000 bp Marker之間;pcDNA3-HERG被切成很接近的兩條目的條帶,瓊脂糖電泳如圖1所示,與預計大小相同。將pcDNA3-HERG的小片段基因條帶和pEGFP-N1端基因條帶通過T4 DNA連接酶進行基因融合,重組成pEGFP-N1-HERG綠色熒光真核表達載體。與預期的pEGFP-N1和結合蛋白目的基因片段長度一致(圖1)。
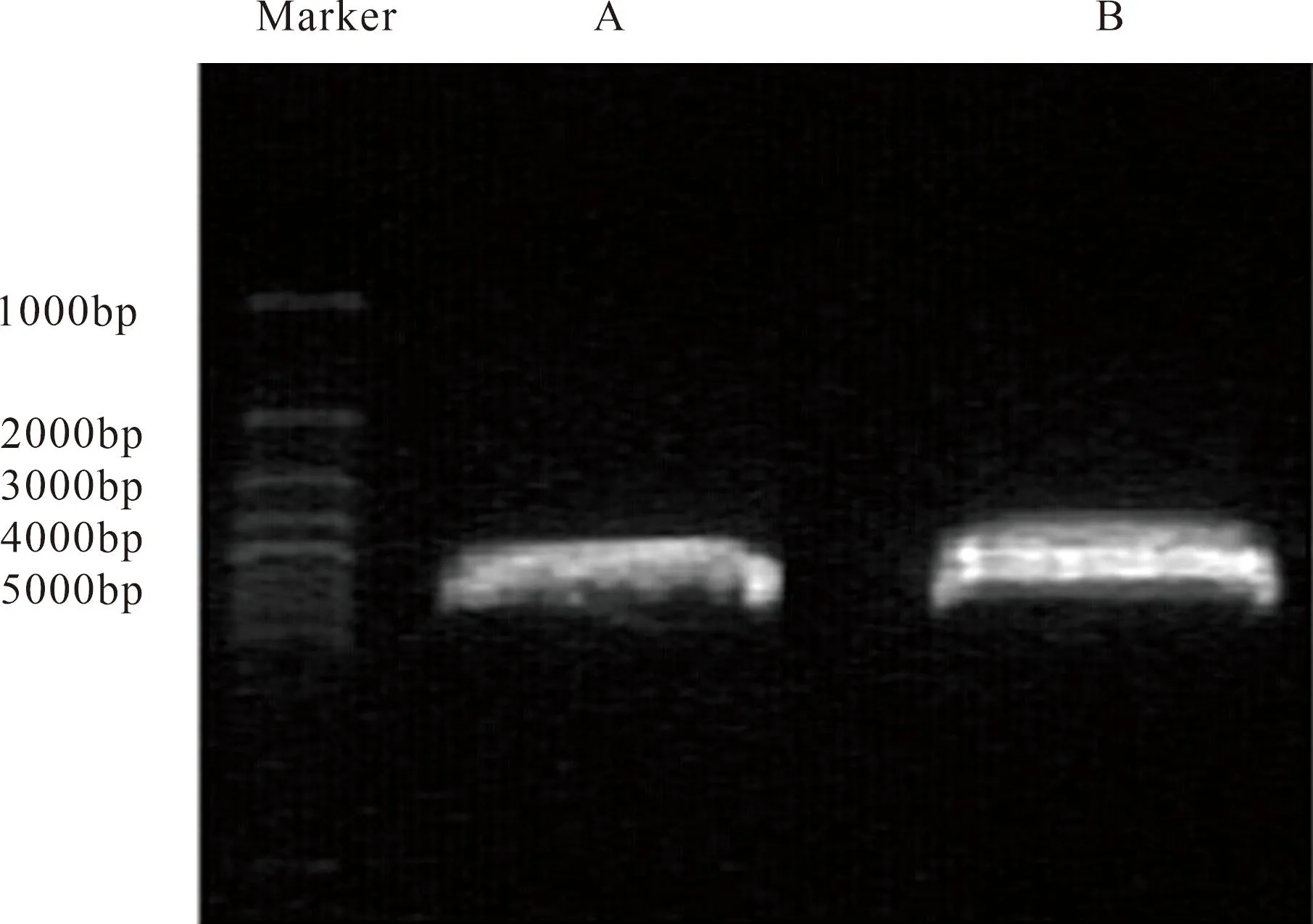
圖1 質粒pcDNA3-HERG和pEGFP-N1的雙酶切電泳圖
2.2 綠色熒光表達載體pEGFP-N1-HERG的鑒定
2.2.1 酶切鑒定:由于目的基因HERG和pEGFP-N1載體基因序列中均有一處BamHⅠ酶切位點,因此重組真核表達載體pEGFP-N1-HERG可被BamH Ⅰ酶切成4000 bp和5000 bp兩條帶,與預計大小相同(圖2)。

圖2 質粒pEGFP-N1-HERG酶切電泳圖
2.2.2 測序分析鑒定:將HERG基因成功連接到pEGFP-N1載體上,pEGFP-N1-HERG測序行雙向測通后的測序結果與美國國立衛生圖書館Genebank中HERG基因堿基序列進行匹配比對,結果示pEGFP-N1-HERG載體中插入的HERG基因堿基序列與Genebank中的序列匹配一致,保證了正常HERG蛋白的表達及通道功能的實現。
2.2.3 熒光顯微鏡觀察細胞pEGFP-N1-HERG表達:pEGFP-N1-HERG轉染后熒光顯微鏡下可見融合有綠色熒光蛋白的目的蛋白在HEK293T細胞中的表達(圖3)。

圖3 轉染后HEK293T細胞熒光表達(脂質體染色,×10)
2.3 EGFP表達的流式細胞儀分析 流式細胞儀分析表明三組轉染質粒pEGFP-N1-HERG的HEK293T細胞經不同糖濃度(5、17.5、30 mmol/L)干預后EGFP轉染陽性效率分別為(69.29±6.05)%、(68.74±11.37)%和(73.92±12.11)%,轉染效率比較無統計學差異(均P>0.05),而HEK293細胞EGFP相對平均熒光強度分別為209.18±9.99、187.11±11.43和124.47±27.30,比較差異有統計學意義(P< 0.05)。見表1(圖4)。

表1 葡萄糖干預48 h后流式計數pEGFP-N1-HERG的陽性細胞百分比和平均熒光強度

A:葡萄糖5 mmol/L;B:葡萄糖17.5 mmol/L;C:葡萄糖30 mmol/L
3 討 論
HERG基因編碼心肌細胞快速延遲整流電壓門控鉀通道(Kv11.1)α亞單位,是心肌動作電位復極期重要的外向電流,各種因素致Kv11.1通道異常使IKr振幅減低,導致LQTS,從而誘發尖端扭轉性室速(TdP),增加心律失常和猝死的風險[11-13]。并且Kv11.1是多種藥物致QT間期改變甚至誘發心律失常的結合位點,同時也是抗心律失常藥治療的重要靶點區域[14]。
pEGFP-N1是一種小分子量并優化的突變型增強GFP,蛋白大小約26.9kDa,其具有更高的靈敏度和熒光強度,且熒光性質穩定,沒有物種特異性,對細胞的生長和功能無顯著的影響,可行活細胞檢測[15]。近年大量研究廣泛采用將目的基因重建到攜帶熒光蛋白基因的pEGFP-N1表達載體中構建成表達綠色熒光融合目的蛋白細胞模型,在熒光顯微鏡下根據綠色熒光強弱及位置反映目的蛋白表達量及轉運定位,且EGFP對靶基因產物的空間構象和功能沒有影響,它已成為檢測目的基因表達方式的最佳報告基因[16-19]。此目的蛋白自帶增強熒光的細胞模型在研究蛋白表達及定位較免疫熒光法更加方便快捷,簡化和節省了免疫熒光后期的抗體孵育及顯色時間;還可在不需要額外熒光抗體標記的情況下快速進行精確的流式細胞學研究[20]。
QT間期異常延長是糖尿病(DM)患者最顯著的心電障礙,同時LQTS患者也表現出更高的糖尿病負荷。眾所周知,hERG編碼快速延遲IKr,HERG通道易受遺傳缺陷和環境因素的影響,在大多數情況下會導致HERG功能的抑制[21]。事實上,大多數長QT綜合征的病例都歸因于HERG通道的功能障礙,尤其是治療藥物引起的功能障礙[22-23]。可以推測,HERG的改變也可能參與了高血糖誘導的QT延長,在糖尿病心肌病中,hERG通道的表達嚴重下調,這種下調是減慢復極和延長QT間期的關鍵因素。本研究發現在高糖處理的hERG-hek細胞中,hERG蛋白的表達隨高糖濃度依賴性降低,和SHI研究結果一致[24]。既往研究發現高糖通過影響Hsp90的表達及其與hERG的相互作用抑制hERG通道的生成。此外,高糖誘導的hERG通道抑制可以通過上調激活轉錄因子-6和內質網伴侶蛋白calnexin的表達水平來激活未折疊蛋白應答,表明抑制非成熟hERG蛋白轉運是糖尿病心肌病中hERG缺乏的潛在機制[24]。同時還發現在胰島素存在下,由高糖引起的Hsp90的抑制顯著恢復,Hsp90的表達增加,這些結果表明胰島素通過降糖可以上調hERG通道的表達,并拯救高糖引起的hERG通道蛋白缺失。還有研究報道高糖誘導的hERG通道缺乏是由于抑制了蛋白在內質網的折疊和轉運,此外也證實胰島素可促進hERG通道的表達,改善高糖誘導的hERG通道抑制的潛在機制[25]。還有些研究報道hERG蛋白表達減少是由氧化應激引起的,可能由高糖刺激導致的活性氧自由基(ROS)參與的氧化應激所致,而胰島素具有抗氧化特性并降低細胞內ROS水平[25]。此外,胰島素通過其抗氧化作用改善糖尿病模型家兔IKr/hERG功能,該研究也證實胰島素顯著增加了Kv11.1蛋白和Ikr電流的表達。此外,高糖誘導的Kv11.1蛋白和Ikr電流的降低可以通過胰島素來改善,源于胰島素可抑制高糖條件下Kv11.1通道蛋白表達減少的作用。上述研究均證實高糖誘導的Kv11.1功能障礙是可預防和可逆的,而胰島素在治療這些糖尿病患者心電問題中是非常有效的。
本研究證明在高糖處理的hERG-HEK293T細胞中,hERG蛋白表達隨高糖濃度依賴性降低。既往研究發現高糖通過抑制Hsp90和hERG/Hsp90復合物的表達,誘導hERG通道轉運缺陷,隨后,內質網應激反應和增加的蛋白質降解在高糖處理的hERG-HEK293細胞中被激活。此外,研究還證實應用胰島素可以恢復hERG蛋白的表達和hERG電流。這些發現表明,抑制轉運是糖尿病心肌病中hERG缺乏的潛在機制。
綜上所述,本研究證實持續高糖狀態通過抑制hERG的表達導致Kv11.1蛋白生成減少,致使IKr門控離子通道功能異常,導致K+外流減少,復極時間延長,在心電圖上表現為QT間期延長,易誘發尖端扭轉型室性心動過速。這一發現闡明了糖尿病誘導的hERG通道表達缺陷的一種細胞內機制。這些發現將極大地改善目前對糖尿病心肌病中hERG蛋白表達降低的認識,為進一步研究高糖在hERG細胞中的作用奠定了基礎,并可能為糖尿病患者的長期高糖狀態下QT間期延長提供有效的研究基礎。
猜你喜歡
人人健康(2023年26期)2023-12-07 03:55:46
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
中國生殖健康(2019年2期)2019-08-23 08:12:10
中國衛生標準管理(2015年1期)2016-01-14 03:41:27
藥學與臨床研究(2015年4期)2015-06-05 11:35:51
