基于石墨烯泡沫的電增強固相微萃取-高效液相色譜法測定魚肉中3 種磺胺類的殘留
2024-04-08 02:28:22孫瑞雪方禹文陳冬妍陳全勝陳曉梅
食品科學 2024年5期
孫瑞雪,方禹文,陳冬妍,陳全勝,陳曉梅
(集美大學海洋食品與生物工程學院,福建 廈門 361021)
磺胺類抗生素(sulfonamides,SAs)是一類廣譜抗菌藥物,其結(jié)構(gòu)以對氨基苯磺酰胺為主,SAs的基本化學結(jié)構(gòu)式如圖1所示。在水產(chǎn)品養(yǎng)殖中,SAs由于具有化學性質(zhì)穩(wěn)定、抗菌譜廣、價格便宜等優(yōu)點而得到廣泛應用[1]。近年來,隨著水產(chǎn)品集約化養(yǎng)殖規(guī)模的不斷擴大,部分養(yǎng)殖戶為減少高密度水產(chǎn)品的養(yǎng)殖病害,超量、超期使用SAs,造成水產(chǎn)品中較高的SAs殘留。消費者長期攝入此類水產(chǎn)品,將導致SAs在體內(nèi)蓄積,損傷人體免疫系統(tǒng),引起胃腸道疾病,甚至出現(xiàn)休克和內(nèi)臟損害[2]。為保護消費者的健康,中國、歐盟等多個國家和地區(qū)規(guī)定,在食用動物組織中添加SAs的最大限量為100 μg/kg[3]。但是,水產(chǎn)品中SAs殘留超標的事件還是頻繁出現(xiàn)。SAs的傳統(tǒng)檢測方法以高效液相色譜(high performance liquid chromatography,HPLC)法為主,但HPLC法的前處理過程比較復雜、耗時較長,不利于對基質(zhì)復雜的水產(chǎn)品中SAs殘留的檢測。目前廣泛使用的前處理方法有液液萃取和固相萃取等,這些前處理方法普遍存在程序比較復雜、有機溶劑的高消耗、耗時、高環(huán)境污染、自動化難度大、成本高和吸附劑選擇少等缺點。因此,開發(fā)一種高效、簡便和環(huán)保的前處理方法,對于實現(xiàn)SAs的簡便、高精度檢測,具有很大的現(xiàn)實意義。

圖1 SAs的基本化學結(jié)構(gòu)Fig.1 Basic chemical structure of SAs
固相微萃取(solid phase microextraction,SPME)技術(shù)是在固相萃取[4]技術(shù)的基礎(chǔ)之上發(fā)展而來的,具有簡單、高效和無毒害等特點,被廣泛應用于樣品前處理中。SPME主要有纖維萃取和管內(nèi)萃取兩種模式。纖維萃取模式常見的有直接萃取[5-7]、頂空萃取[8-10]和膜保護萃取[11-12]。纖維萃取裝置組成比較簡單,包括萃取頭和手柄,與注射器相似。纖維萃取模式的基體支持物主要是石英纖維、不銹鋼絲等材料,將固定相涂層涂漬在支持物表面,然后對樣品中的待測物進行萃取和富集。管內(nèi)萃取則以石英毛細管[13-15]和peek管[16-17]等材料作為基體支持物。SPME的富集能力主要取決于涂層材料的吸附性能、極性和厚度等,而上述兩種萃取模式都存在涂層材料用量少、萃取容量不夠大的缺點,這在一定程度上影響了SPME的富集效率[18]。近年來,整體柱(monolith,M)作為一種新的SPME萃取模式,因其單體豐富、制作工藝簡單、滲透性好、易于功能化等優(yōu)點而受到人們的關(guān)注。此外,隨著性能優(yōu)越的納米材料的不斷引入,M的萃取性能也顯著提升。例如,Wang Yu[19]和Wu Xinze[20]等研究表明,將碳基材料等比表面積大的材料引入M中,可以顯著增加M的比表面積,并豐富M中的官能團,有效提高萃取效率。石墨烯是一種新型的碳質(zhì)材料,具有大比表面積、高韌性和高導電性等優(yōu)點,本課題組在前期研究中用離子液體作為石墨烯的黏合劑,制備了一種具有優(yōu)異導電性和表面易于更新的電極,并成功用于貝類中Pb2+、Cd2+含量的同時檢測[21]。但石墨烯間較強的相互作用致使其易于堆疊,造成其比表面積減小。所以,人們研制出一種三維結(jié)構(gòu)的石墨烯,它可以在維持原有超大比表面積、超高導電率的同時,避免石墨烯的片層間發(fā)生堆疊。本課題組在前期研究中利用水熱法合成了三維石墨烯并成功應用蝦加工副產(chǎn)物中Hg2+的脫除[22]。氨基功能化的三維石墨烯泡沫(nitrogen-doped three-dimensional graphene foam,NGF)具有比表面積大、孔徑豐富且規(guī)則、導電性好等優(yōu)點。本研究利用NGF功能化M(M@NGF),一方面可提高M的比表面積,增強萃取效果;另一方面可提高M的導電性,有利于將電化學技術(shù)引入SPME中,提高萃取效率。目前鮮有研究將NGF用于功能化M,且鮮有利用M萃取SAs的報道。此外,傳統(tǒng)的SPME法主要基于被動分配萃取目標物,萃取速度不夠理想。近年來,科研人員開發(fā)出電輔助增強固相微萃取(electroenhanced solid phase microextraction,EE-SPME)技術(shù),在萃取過程中施加電場,帶電的待測物可以通過電泳被捕獲到固定相中,此方法的萃取速度明顯比被動分配要快[23-24]。
本研究以檢測魚肉中磺胺噻唑(sulfathiazole,ST)、磺胺二甲基嘧啶(sulfamethazine,SM2)和磺胺二甲氧嘧啶(sulfadimethoxypyrimidine,SMM)3 種物質(zhì)為目的,用離子液體1-乙烯基-3-辛基咪唑四氟硼酸鹽(1-octyl-3-vinylimidazolium tetrafluoroborate,VOT)作為功能單體,2,2′-偶氮二異丁腈(2,2′-azobisisobutyronitrile,AIBN)作為熱引發(fā)劑,二乙烯基苯(divinylbenzene,DVB)作為交聯(lián)劑,二甲基亞砜(dimethyl sulfoxide,DMSO)作為致孔劑,NGF作為導電增強劑,通過超聲、裝管、成型,得到穩(wěn)定性良好、孔徑豐富的M。采用EE-SPME法結(jié)合HPLC技術(shù),實現(xiàn)對SAs快速、準確的測定。最后,將本方法應用于草魚、羅非魚、金鯧魚、鱸魚4 種魚肉中ST、SM2、SMM含量的檢測。綜上所述,本研究基于原位聚合法制備了M@NGF,并將EE-SPME和HPLC結(jié)合,開發(fā)了一種快速、靈敏的檢測魚肉中SAs的方法,為食品中抗生素的檢測提供了新的方法參考。
1 材料與方法
1.1 材料與試劑
魚肉樣品(草魚、羅非魚、金鯧魚、鱸魚)購自廈門市集美區(qū)水產(chǎn)批發(fā)市場。
磺胺類標準品:ST(GBW(E)060904,99.9%)、SM2(71169,99.9%)、SMM(190-11101,99.0%)壇墨質(zhì)檢-國家標準物質(zhì)中心;VOT(99%)中國科學院蘭州化學物理研究所;乙腈(色譜純)、甲醇(色譜純)美國天地試劑有限公司;DVB(80%)、DMSO(98%)、AIBN(97%)、γ-聚谷氨酸(γ-polyglutamic acid,γ-PGA)、無水乙醇、甲酸(色譜純)麥克林化工有限公司;氧化石墨烯(graphene oxide,GO)分散液(2 mg/mL)蘇州碳豐科技有限公司;不銹鋼絲(stainless steel wires,SSW)上海璽帛金屬制品有限公司。
每周用乙腈配制ST、SM2、SMM原液(100 μg/mL),4 ℃保存。標準工作溶液在使用前要用乙腈稀釋。
1.2 儀器與設備
Sigma 300掃描電子顯微鏡(scanning electron microscope,SEM)德國卡爾蔡司股份公司;1260高效液相色譜系統(tǒng)(配備2998可變波長檢測器(variable wavelength detector,VWD))美國安捷倫公司;KH-250E超聲波清洗機 昆山禾創(chuàng)超聲儀器有限公司;DHG-9053A干燥箱 上海一恒科學儀器有限公司;Unique-R20+UV超純水機 廈門銳思捷水純化技術(shù)有限公司;PS-305DM直流電源 深圳市邦企創(chuàng)源科技有限公司;RCT basic磁力攪拌器 德國艾卡有限公司;pH700型pH計 美國優(yōu)特儀器有限公司;ALPHA傅里葉變換紅外光譜(Fourier transform infrared,F(xiàn)TIR)儀德國布魯克儀器公司;ASAP2460全自動比表面及孔隙度(BET)分析儀 美國麥克儀器公司。
1.3 方法
1.3.1 NGF的制備
將20 mg的γ-PGA加入到10 mL的GO中,攪拌2 h至充分溶解。然后轉(zhuǎn)移到20 mL的反應釜中,90 ℃水熱反應3 h,得到NGF凝膠,再用乙醇和去離子水依次清洗,最后真空冷凍干燥2 d,即得到所需NGF[25]。
1.3.2 M@NGF的制備
根據(jù)原位聚合的方法制備多孔M@NGF[26]。具體如下:首先,稱取130 mg功能單體(VOT)、100 mg交聯(lián)劑(DVB)和5 mg熱引發(fā)劑(AIBN),溶解于100 mg致孔劑(DMSO)中,然后加入5 mg NGF,超聲5 min使其充分混合;然后,將混合溶液注入長為3 cm、內(nèi)徑為2 mm的模具管中,聚合物溶液高度控制在2 cm左右。將管朝下的一端用硅膠塞密封,另一端垂直插入一根長度為10 cm的SSW;最后,在70 ℃的烘箱中進行原位聚合16 h。聚合完成后,冷卻至室溫,脫去外層模具管,即得到M@NGF。將M@NGF浸泡在甲醇中,以清除未反應的組分。
1.3.3 M@NGF的表征
1.3.3.1 紅外光譜表征
將溴化鉀粉末與M@NGF按照質(zhì)量比1∶100的比例稱量后置于研缽中充分研磨,用壓片機壓成均勻薄片,放于FTIR儀中,測定其紅外吸收光譜。
1.3.3.2 比表面積和孔徑分布的測定
使用全自動比表面積分析儀測量樣品的比表面積和孔徑分布。測定前,將M@NGF樣品在90 ℃條件下真空脫氣8 h。待樣品冷卻至室溫后,放入全自動比表面積和分析儀中,以高純氮為介質(zhì)進行吸附。采用多點BET方程計算比表面積,使用BJH方法計算孔徑分布。
1.3.3.3 SEM表征
將待測樣品用導電膠固定在樣品盤上,并對試樣進行噴金處理,在SEM下觀察樣品結(jié)構(gòu)。
1.3.4 EE-SPME萃取SAs的過程
EE-SPME萃取SAs的過程如圖2所示。首先,將M@NGF依次浸入甲醇和超純水中10 min以活化M@NGF。然后,將M@NGF和SSW插入溶液中,分別連接直流電源負極和正極,施加-1.0 V的電壓,在300 r/min條件下對含有SAs的溶液萃取35 min。萃取完成后,將M@NGF和SSW移到1 mL的混合洗脫溶液(甲醇/水和甲酸/水)中,分別連接電源的正極和負極,施加1.5 V的電壓,在300 r/min的攪拌速度下解吸SAs。20 min后,用N2將解吸液吹干,再用1000 μL的初始流動相重新溶解干燥的殘渣,最后用0.22 μm的濾膜過濾,濾液用HPLC分析檢測。
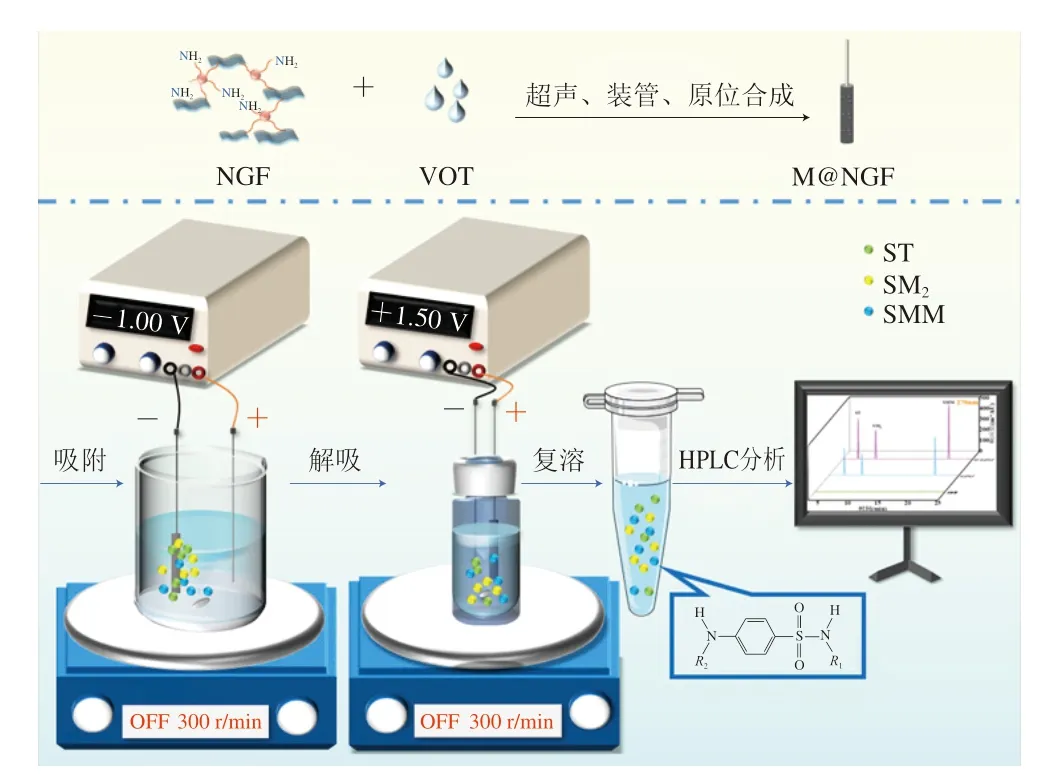
圖2 基于M@NGF的EE-SPME HPLC法檢測SAs的示意圖Fig.2 Schematic diagram of M@NGF based EE-SPME HPLC method for the detection of SAs
1.3.5 樣品制備
將準確稱量的1 g均質(zhì)魚肉樣品加入離心管中,隨后加入50 mL 30%甲醇水混合溶液,渦旋振蕩5 min,超聲處理10 min。然后將混合物在5000 r/min離心10 min。最后將上清液轉(zhuǎn)移到燒杯中,進行1.3.4節(jié)中的操作。
1.3.6 色譜條件
色譜柱:Welch C18反相色譜柱(250 mm×4.6 mm,5 μm);流動相:0.1%甲酸溶液(A相)和乙腈(B相);流速1 mL/min,柱溫35 ℃,進樣量80 μL。梯度洗脫程序如表1所示。此方法用保留時間確定SAs,用色譜峰面積量化和計算萃取回收率。

表1 SAs的梯度洗脫程序Table 1 Gradient elution procedures for analysis of SAs
1.3.7 萃取條件的優(yōu)化
對吸附時間(20~40 min)、攪拌速度(100~400 r/min)、吸附電壓(-2.5~-0.5 V)、離子強度(0%~0.8%)、樣品溶液pH值(2~10)、解吸時間(5~25 min)、解吸電壓(1.0~3.0 V)和解吸液組成(甲醇、甲酸和水的體積分數(shù))進行優(yōu)化。
1.3.8 萃取性能
分別從M@NGF的富集效果、萃取選擇性、使用壽命和檢測結(jié)果的重現(xiàn)性4 個方面評價M@NGF的萃取性能。
1.4 數(shù)據(jù)處理與分析
數(shù)據(jù)處理分析采用軟件Origin 2021、Excel和PowerPoint,其中作圖使用Origin 2021和PowerPoint,線性回歸方程、偏差值、回收率采用Excel處理數(shù)據(jù)。
2 結(jié)果與分析
2.1 M@NGF的表征
首先用傅里葉變換紅外光譜對M@NGF進行了表征,圖譜如圖3A所示。在M@NGF的紅外曲線中,位于1640 cm-1和1058 cm-1處的吸收峰分別屬于VOT的咪唑基和BF4-基團的吸收。在3149、1601、1554 cm-1和1456 cm-1處的吸收峰可以證明苯環(huán)的存在。3410 cm-1處的吸收峰為N—H的伸縮振動峰,1160、1596、2357 cm-1處的吸收峰則分別為NGF的C—O—C伸縮振動、N—H彎曲振動和C—N的耦合以及C—N的對稱伸縮振動。以上結(jié)果表明,M@NGF的紅外曲線中包含了VOT和NGF的特征吸收峰,說明NGF成功對M進行了功能化。M@NGF的BET曲線如圖3B所示,M@NGF表現(xiàn)為II型等溫線,孔徑約為40 nm,經(jīng)過計算得出M@NGF的比表面積約為25 m2/g。
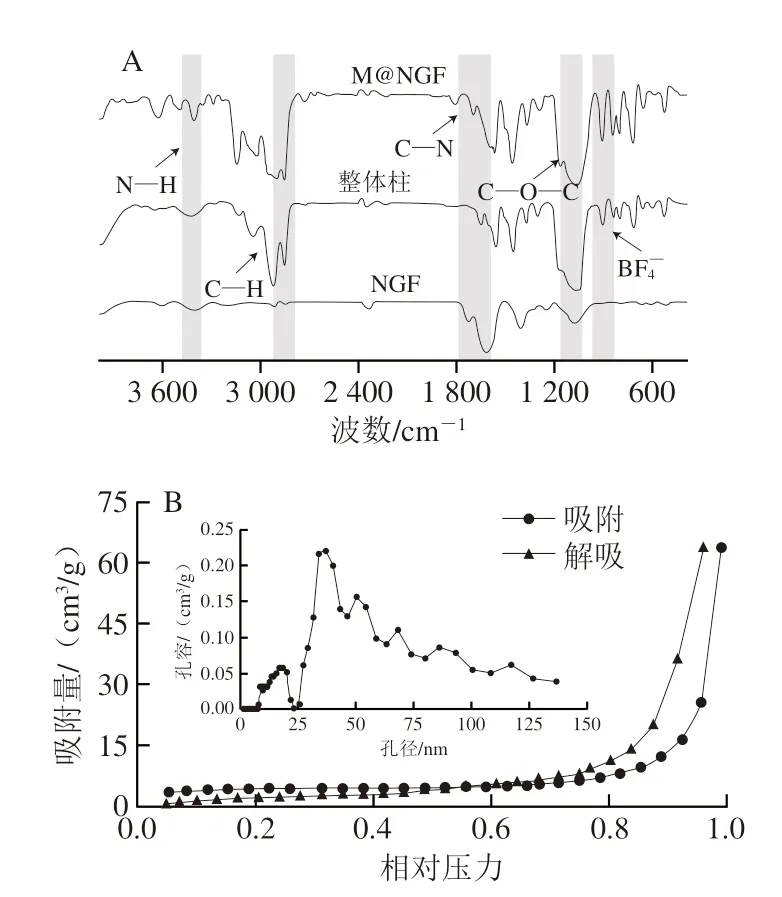
圖3 NGF、整體柱以及M@NGF的FTIR曲線(A)和M@NGF的BET曲線(B)Fig.3 FTIR spectra of NGF,monolith,and M@NGF (A) and BET curves of M@NGF (B)
NGF的SEM圖如圖4A所示,可以看出NGF呈片狀堆疊狀,且具有較多孔洞,孔徑約為10 μm左右。由圖4B中M@NGF的SEM圖可以看出,SSW的表面包裹著均勻的聚合物,較為平整、光滑;由圖4B的插圖中M@NGF的照片可以看出其長度約為2 cm;圖4C1為NGF的橫截面SEM圖,可以看出M@NGF內(nèi)徑約為2 mm。圖4C2為其放大的SEM圖,可以看出聚合物內(nèi)部孔洞較為豐富且均勻。這些特殊的多孔結(jié)構(gòu)可以為液體流動提供較大的孔道,有利于加快待測物對流傳質(zhì),進而提高對SAs的萃取效率。

圖4 NGF(A)、M@NGF(B)和M@NGF截面(C)的SEM圖Fig.4 SEM images of NGF(A),M@NGF (B) and cross-section of M@NGF (C)
2.2 吸附和解吸條件的優(yōu)化
為獲得對SAs的最佳萃取效率,實驗優(yōu)化了EE-SPME過程的條件,包括吸附和解吸電壓、吸附和解吸時間、樣品溶液的pH值、攪拌速度、解吸液組成及含水量等。
2.2.1 吸附時間、攪拌速度、離子強度和pH值的影響
2.2.1.1 吸附時間的影響
SPME是基于待測物在樣品及萃取相之間平衡分配的一個過程,但EE-SPME在電場的作用下,可以有效加快吸附速率。本研究考察了在外加電場的作用下,萃取量在20~40 min范圍內(nèi)的變化。結(jié)果如圖5A所示(E表示施加電壓,下同),所有目標物的萃取量在20~35 min內(nèi)迅速增加,在35~40 min內(nèi)基本保持不變。此外,在其他因素保持不變的情況下,以不施加電場的SPME的吸附速率作為對照。可以看出,隨著吸附時間的延長,除了ST以外,SM2和SMM的萃取量都有所增加。在35~40 min之間,不施加電場的SPME,對3 種SAs的萃取量仍有明顯增加的趨勢,說明還未達到吸附平衡。此外,在35 min時,未加電場的SPME對3 種SAs的萃取效果遠差于EE-SPME的效果,說明EE-SPME方法可以顯著增強對3 種SAs的萃取效率,并且在35 min時便可達到吸附平衡。
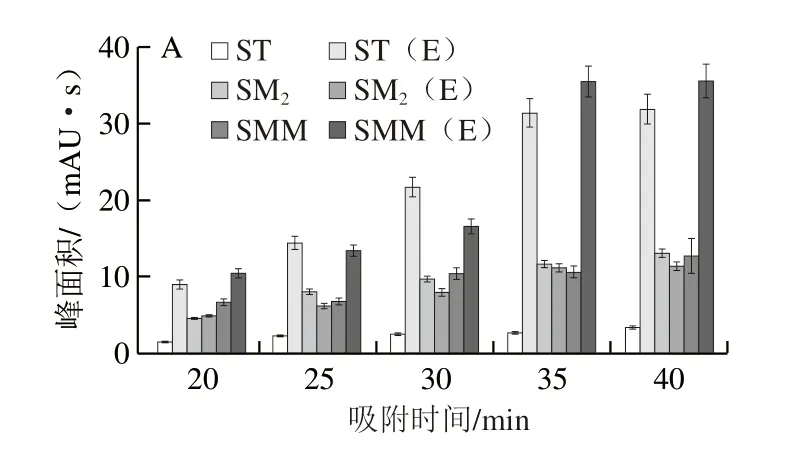
圖5 吸附時間(A)、攪拌速度(B)、離子強度(C)和pH值(D)對萃取效率的影響Fig.5 Effects of adsorption time (A),agitation speed (B),ionic strength (C)and pH (D) on the extraction efficiency
2.2.1.2 攪拌速度的影響
考察攪拌速度在100~400 r/min范圍內(nèi)對萃取效果的影響,結(jié)果如圖5B所示。在100~400 r/min范圍內(nèi),300 r/min的萃取效果最好。攪拌是為了提高目標物的溶解效率,使萃取過程中的目標物均勻分散在樣品溶液中,本實驗采用了電壓作為輔助萃取手段,當攪拌速度小于300 r/min時,溶液分子擴散到電極表面速率較慢,無法在電極表面進行有效傳質(zhì),而當攪拌速度太高時,電極表面的目標物分子擴散太快,擴散層很薄,目標物來不及被電極上的吸附劑吸附,導致信號降低,且攪拌速度過快,不利于電極表面吸附劑的穩(wěn)定性,所以本實驗選取300 r/min作為后續(xù)實驗的攪拌速度。
2.2.1.3 離子強度的影響
通過添加質(zhì)量分數(shù)為0%~0.8%的NaCl,研究離子強度對萃取量的影響。如圖5C所示,在質(zhì)量分數(shù)為0%~0.6%時,SAs的峰面積隨著NaCl含量的增加而上升;而在NaCl質(zhì)量分數(shù)大于0.6%時,峰面積呈現(xiàn)下降的趨勢。這可能是由于少量的NaCl可以提高溶液的導電率,從而有利于分析物的傳質(zhì);然而,過量的Cl-在電場的作用下可能會與電離的SAs競爭,與吸附劑結(jié)合,從而不利于SAs的萃取。基于此結(jié)果,后續(xù)研究選擇添加0.6%的NaCl提升萃取性能。
2.2.1.4 樣品溶液pH值的影響
因為吸附劑和目標物中存在極性基團,所以樣品溶液的pH值也會影響吸附劑對目標物的萃取性能。所以,本實驗中通過控制pH值在2.0~10.0之間驗證溶液的pH值對萃取效果的影響。如圖5D所示,在pH值為6.0時,M@NGF對于SAs的萃取效果最好。而在pH值小于6.0時,M@NGF和SAs中的N原子發(fā)生了質(zhì)子化,吸附劑與目標物之間會產(chǎn)生靜電排斥,不利于SAs的吸附。隨著溶液pH值的增加,靜電斥力會減弱,同時,氫鍵相互作用也會參與萃取過程,使萃取效果提升。但溶液的pH值太大,SAs的氨基可能會發(fā)生脫質(zhì)子化反應,電場的增強作用減弱,這會導致萃取性能下降[24,27-28]。根據(jù)優(yōu)化的結(jié)果,后續(xù)研究將樣品溶液的pH值控制在6.0。
2.2.2 電壓的影響
吸附和解吸時所施加的電壓是影響萃取效果的重要要素。本實驗考察了吸附和解吸電壓對SAs萃取效果的影響,結(jié)果如圖6所示。當吸附電壓為-1.0 V時,3 種SAs均能達到最大的峰面積(圖6A)。在解吸步驟中(圖6B),電壓在1.0~1.5 V范圍內(nèi),3 種SAs的峰面積都隨著電壓的升高而增加,而在1.5~3.0 V范圍內(nèi)的解吸效果明顯變差。根據(jù)此結(jié)果,將吸附和解吸步驟的外加電壓分別設置為-1.0 V和1.5 V。

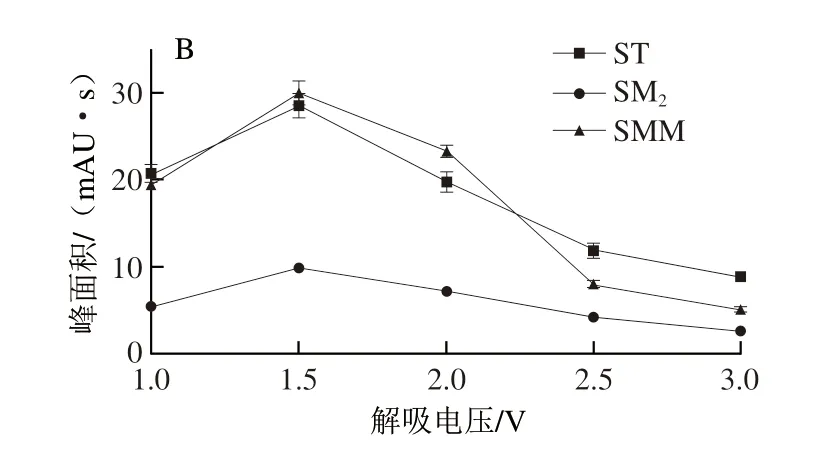
圖6 吸附電壓(A)和解吸電壓(B)對萃取效率的影響Fig.6 Effects of adsorption voltage (A) and desorption voltage (B) on the extraction efficiency
2.2.3 解吸時間和解吸液的影響
2.2.3.1 解吸時間的影響
圖7A顯示了在5~25 min范圍內(nèi)解吸時間對解吸效果的影響。在電場作用下,20 min內(nèi)幾乎可以完全釋放出M@NGF中所萃取的SAs。而在未施加電壓的實驗中,可以觀察到SAs有緩慢的解吸過程,但即使解吸時間延長到25 min,仍不能完全解吸出SAs。以上結(jié)果表明,在萃取過程中施加電場可以明顯加快吸附和解吸速度,縮短樣品制備的時間。因此,在后續(xù)實驗中,選擇20 min作為解吸時間。

圖7 解吸時間(A)、甲醇體積分數(shù)(B)、甲酸體積分數(shù)(C)和水體積分數(shù)(D)對萃取效率的影響Fig.7 Effect of desorption time (A),and volume fractions of methanol (B),formic acid (C) and water (D) on the extraction efficiency
2.2.3.2 甲醇體積分數(shù)的影響
考察了解吸液中甲醇體積分數(shù)為80%~100%時對萃取效果的影響。結(jié)果如圖7B所示,當甲醇體積分數(shù)為90%時,表現(xiàn)出最好的解吸效果。
2.2.3.3 甲酸體積分數(shù)的影響
由于SAs的吸附中存在B-N配位、離子交換和氫鍵作用等,所以在洗脫液中加入一些酸有利于破壞相關(guān)的相互作用。基于此,在甲醇(90%)溶液中加入不同比例的甲酸,以洗脫M@NGF中吸附的SAs。如圖7C所示,2%的甲酸對SAs的洗脫效果最佳。基于上述結(jié)果,后續(xù)研究選擇2%的甲酸與90%的甲醇的混合溶液作為洗脫液。
2.2.3.4 水體積分數(shù)的影響
已有研究表明,改變解吸溶劑中的水體積會改變洗脫液的導電性,從而影響解吸的效果。如圖7D所示,當洗脫液中水的體積分數(shù)由5%增加到10%時,3 種SAs的峰面積均明顯增加,此后便隨著水體積的增加而減少。這也證實了解吸溶劑中有少量的水存在時可以提高吸附劑的導電性,一定程度上促進了吸附劑中SAs的釋放。但當洗脫溶液含水量過多時,甲醇和甲酸在洗脫液中的比例會相應降低,反而導致吸附的SAs不能完全從M@NGF中釋放。綜合考慮,在后續(xù)研究的中,解吸液中水的體積分數(shù)設置為10%。
2.3 M@NGF的萃取性能
考察了基于M@NGF的EE-SPME和直接SPME對SAs的萃取效果。如圖8A所示,直接用樣品溶液進樣只能出現(xiàn)很弱的色譜峰(曲線a),而經(jīng)過EE-SPME(曲線c)后,SAs的響應信號增大了58~74 倍(基于M@NGF的EE-SPME法對ST、SM2和SMM的富集倍數(shù)分別為74、58和64),同時與未施加電場的直接SPME(曲線b)相比,SAs的響應信號也明顯增強。以上結(jié)果表明,在萃取過程中施加電場可以提高M@NGF對SAs的萃取性能。
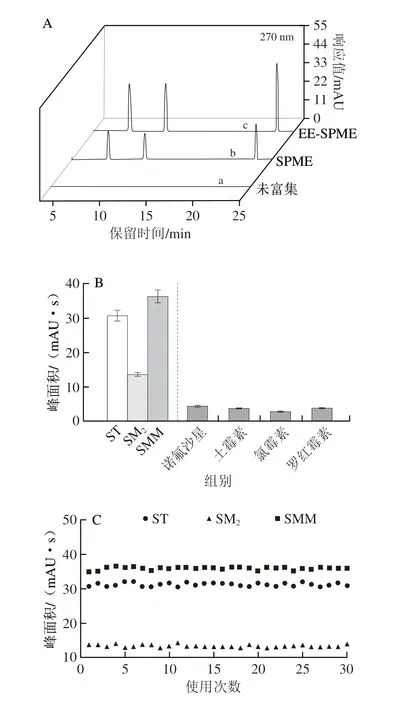
圖8 不同前處理的SAs色譜圖(A)和M@NGF的選擇性(B)、使用壽命(C)和重復性(D)Fig.8 Chromatograms of SAs (A) after different pre-treatments and selectivity (B),lifetime (C) and repeatability (D) of M@NGF
考察了M@NGF對SAs的選擇性。選取諾氟沙星、土霉素、氯霉素、羅紅霉素作為干擾物(含量均為50 μg/kg),研究M@NGF對SAs選擇性。如圖8B所示,在4 種抗生素存在的情況下,M@NGF對SAs的萃取仍然能保持原有效果。這表明本實驗所制備的M@NGF對SAs具有較好的選擇性。由于物理吸附中的靜電作用與物質(zhì)的Zeta電位和pH值以及pKa(酸度系數(shù))相關(guān),pKa會影響分子的存在形式從而影響吸附效果,本實驗選取的4 種干擾物的pKa普遍高于SAs的pKa。本實驗所選取的pH值為6,通過查閱文獻,諾氟沙星在pH<6.1時以陽離子存在,不利于被吸附[29];土霉素的最佳吸附pH值為8[30];氯霉素在pH值為6時被吸附量最低[31];羅紅霉素在pH 7.2時才能達到最大吸附[32];所以,在pH值為6時,均不利于另外4 種抗生素的吸附,因此,在優(yōu)化好的條件下,本實驗對SAs表現(xiàn)出最好的吸附行為。
考察了M@NGF的使用壽命,用同一根M@NGF連續(xù)萃取30 次后,得到的結(jié)果如圖8C所示,M@NGF的萃取性能沒有明顯變化,相對標準偏差(relative standard deviation,RSD)范圍為1.3%~3.0%,說明M@NGF具有良好的穩(wěn)定性和較長的使用壽命。
考察了M@NGF的重復性,在優(yōu)化的萃取條件下,選擇平行制備的5 根M@NGF對SAs進行萃取、檢測。結(jié)果如圖8D所示,5 根M@NGF萃取到的SAs量無顯著差異,RSD為5.7%,表明M@NGF具有較好的重復性。
2.4 線性范圍和檢出限
根據(jù)上述實驗結(jié)果,最佳萃取條件:吸附時間35 min、吸附電壓-1.0 V、吸附液pH 6、解吸時間20 min、解吸電壓1.5 V、甲醇體積分數(shù)90%、甲酸體積分數(shù)2%。在優(yōu)化的萃取條件下,對5~5000 μg/kg系列含量的SAs用基于M@NGF的EE-SPME、基于M@NGF的直接SPME以及未摻雜NGF的EE-SPME進行萃取、測定。以峰面積y對SAs的含量x繪制工作曲線,基于M@NGF的EE-SPME,ST、SM2和SMM的檢出限分別為1.78、3.16 μg/kg和1.84 μg/kg,定量限均為5 μg/kg,決定系數(shù)(R2)均大于0.9990,結(jié)果如表2所示。基于M@NGF的EE-SPME相對于基于M@NGF的直接SPME和基于未摻雜NGF的EE-SPME兩種方式不管是在檢測范圍還是檢出限方面均表現(xiàn)出更好的檢測性能,說明所建立的基于M@NGF的EE-SPME方法可以在前處理中有效提高SAs的萃取效率,實現(xiàn)對SAs的高效、快速測定。
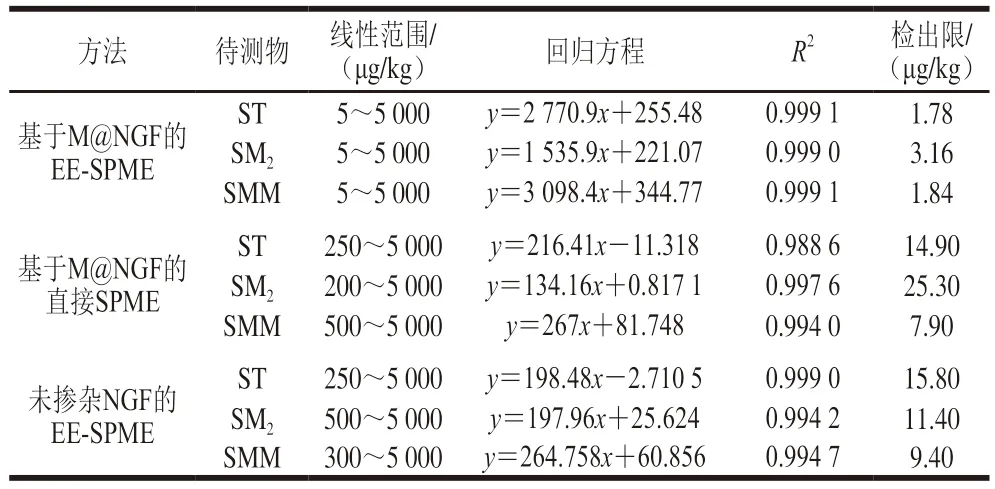
表2 基于M@NGF的EE-SPME和直接SPME以及未摻雜NGF的EE-SPME對SAs的線性范圍和檢出限Table 2 Linear ranges and detection limits of SAs using EE-SPME and direct SPME based on M@NGF and non-NGF-doped EE-SPME
2.5 實際樣品分析和回收率的測定
選取草魚、羅非魚、金鯧魚、鱸魚4 種市場銷售的魚進行分析檢測,均未檢出ST、SM2和SMM。
為考察食品基質(zhì)對萃取效果的影響,選取草魚、羅非魚、金鯧魚、鱸魚4 種魚進行加標回收實驗并計算回收率,結(jié)果如表3所示,3 種SAs的加標回收率范圍為79.2%~110.1%(n=5),RSD范圍為1.4%~9.8%,說明此法的精密度和重復性都比較好。
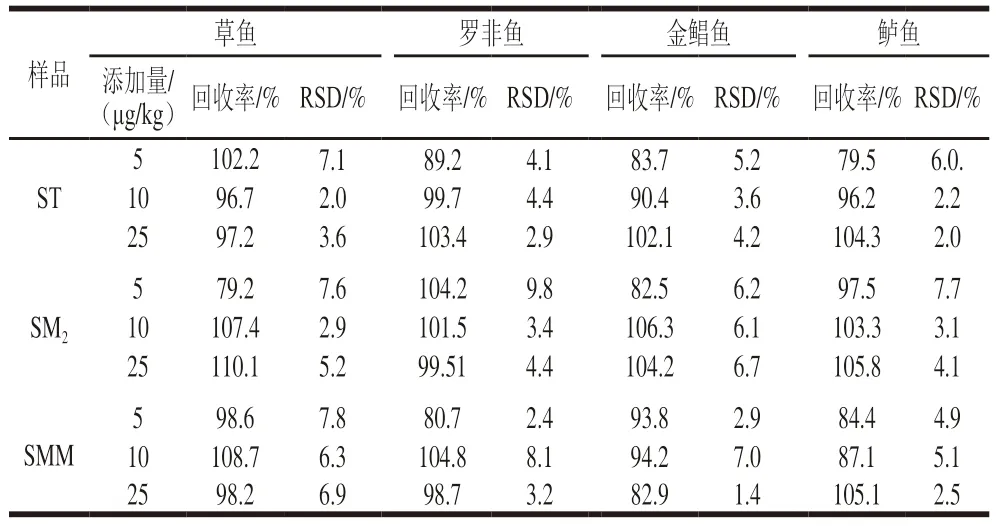
表3 4 種魚肉樣品中3 種SAs的加標回收率Table 3 Recoveries of three SAs in four spiked fish samples
2.6 與其他方法的比較
為了驗證本方法的優(yōu)越性,將此EE-SPME與其他已報道的SPME檢測方法進行比較。結(jié)果如表4所示,所建立的基于M@NGF的EE-SPME方法在檢測SAs時,表現(xiàn)出更寬的檢測范圍和更低的檢出限。相對于傳統(tǒng)的SPME方法,本方法不僅保留了傳統(tǒng)SPME固有的優(yōu)點,還增加了氨基功能化的石墨烯材料和高效的電場驅(qū)動的功能,在增大吸附劑的比表面積的同時還可以有效加快吸附、解吸過程中的傳質(zhì),提高前處理的效率。說明本方法對SAs的檢測具備良好的分析性能。
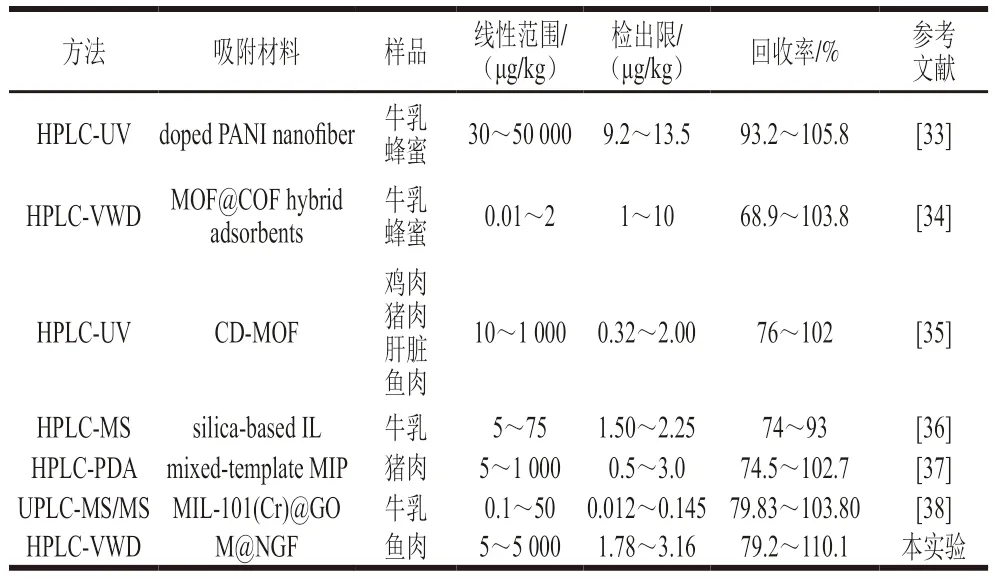
表4 各種 SAs 檢測方法的性能比較Table 4 Comparison of analytical figures of merit of various SA detection methods
3 結(jié)論
本實驗建立了一種基于M@NGF的EE-SPME前處理方法,結(jié)合HPLC法快速測定4 種魚肉中的ST、SM2、SMM殘留。研究結(jié)果表明,所制備的M@NGF不僅能提高SAs的富集效果,還表現(xiàn)出良好的選擇性和穩(wěn)定性。另外,EE-SPME可以有效驅(qū)動電場,提高萃取效率。以吸附時間35 min、吸附電壓-1.0 V、吸附液pH 6、解吸時間20 min、解吸電壓1.5 V、解吸液中含有90%的甲醇和2%的甲酸為最佳萃取條件,本方法對ST、SM2和SMM的檢出限分別為1.78、3.16 μg/kg和1.84 μg/kg,定量限均為5 μg/kg,加標回收率為79.2%~110.1%,RSD范圍為1.4%~9.8%,方法準確度較高,可滿足魚肉中3 種SAs殘留定性、定量分析的要求。本方法集萃取和濃縮為一體,具有過程簡單、快速、重復性較好等優(yōu)點,為魚肉中抗生素殘留安全風險監(jiān)控提供了新的可行途徑。
猜你喜歡
中老年保健(2021年12期)2021-11-30 02:58:01
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
攝影之友(影像視覺)(2019年2期)2019-03-05 08:27:14
中華詩詞(2018年11期)2018-03-26 06:41:34
Coco薇(2016年8期)2016-10-09 02:11:50
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
