新生兒急性心功能不全1例
2024-05-10 01:54:18李淑涓胡黎園張蓉楊琳奚立劉芳曹云周文浩程國強
中國當代兒科雜志 2024年3期
李淑涓 胡黎園 張蓉 楊琳 奚立 劉芳 曹云 周文浩 程國強
(國家兒童醫學中心/復旦大學附屬兒科醫院1.新生兒科;2.內分泌遺傳代謝科;3.心內科,上海 201102)
1 前言
先天性孤立性促腎上腺皮質激素缺乏癥(congenital isolated adrenocorticotropic hormone deficiency, CIAD)是一種罕見的常染色體隱性遺傳病,具有臨床異質性及家族遺傳性,由Steinberg等[1]于1954 年首次報道,該病每年發病率約0.9/100 萬人[2]。CIAD 的特點是繼發性腎上腺皮質功能不全,促腎上腺皮質激素(adrenocorticotropic hormone, ACTH)和皮質醇分泌減少,垂體其他激素分泌正常或短暫性的可逆性升高,無其他結構性垂體缺陷,并排除外源性糖皮質激素應用以及垂體瘤術后所致的ACTH 缺乏[3]。國內關于CIAD病例報道多以成人或兒童為主,新生兒期診斷CIAD的病例報道目前僅有1例[4]。CIAD典型的臨床表現為頑固性低血糖、膽汁淤積和驚厥,而以心功能不全為主要表現的病例暫無報道。CIAD 如未早期識別,可導致新生兒死亡,早期診斷并接受激素替代治療可顯著改善預后。本文描述1 例TBX19基因變異所致以急性心功能不全為主要表現的CIAD患兒的臨床診療經過,以期提高臨床醫生對該病的認識。
2 病例介紹
現病史:患兒男,1 日齡,因生后6 h 出現低血糖伴呼吸暫停收入我院新生兒重癥監護病房。患兒系第5 胎第2 產,胎齡38+6周,順產,出生體重2 840 g,無胎膜早破,羊水清,臍帶、胎盤未見異常。生后Apgar評分1 min 9分,5 min 9分。生后6 h出現呼吸暫停伴四肢抖動,監測經皮血氧飽和度為75%~80%,心率90~100 次/min,末梢血糖0.9 mmol/L,予10%葡萄糖注射液靜脈推注并托背刺激,患兒呼吸無改善,予氣管插管常頻機械輔助通氣、靜脈補液及抗感染治療后,復測末梢血糖3.1 mmol/L,為進一步救治,在氣管插管機械通氣下轉入我院新生兒重癥監護病房。患兒母親正規產檢,孕38+4周因“羊水少”于外院住院保胎治療。患兒父母身體健康,非近親婚配,否認家族遺傳性疾病史。
入院體格檢查:神志清楚,反應欠佳。全身皮膚紅潤,未見色素沉著,無水腫。前囟平軟,口唇紅潤,頸軟。氣管插管常頻機械輔助通氣下,吸氣性凹陷弱陽性,雙肺送氣音對稱。心音有力,律齊,未聞及雜音。腹平軟,肝脾不大,腸鳴音弱。四肢肌張力正常,原始反射可引出,四肢末梢暖,毛細血管充盈時間2 s。
輔助檢查:新生兒基因Panel檢測(生后第21天) 示TBX19基因存在復合雜合突變(NM_005149, exon7: c.917-2A>G 和 NM_005149,exon4:c.608C>T),該基因為CIAD 致病基因[5]。經父母基因驗證,父親攜帶c.917-2A>G,母親攜帶c.608C>T。余實驗室檢查結果見表1。心臟彩超(生后第3 天) 示動脈導管未閉(patent ductus arteriosus, PDA)(3.5 mm),射血分數>60%,二尖瓣輕度反流,三尖瓣輕中度反流。心臟彩超(生后第8天)示左室收縮功能減低(射血分數55%),PDA(2.6 mm),二尖瓣中度反流,三尖瓣中重度反流。肝胰腎上腺超聲、垂體平掃增強磁共振成像檢查未見明顯異常。
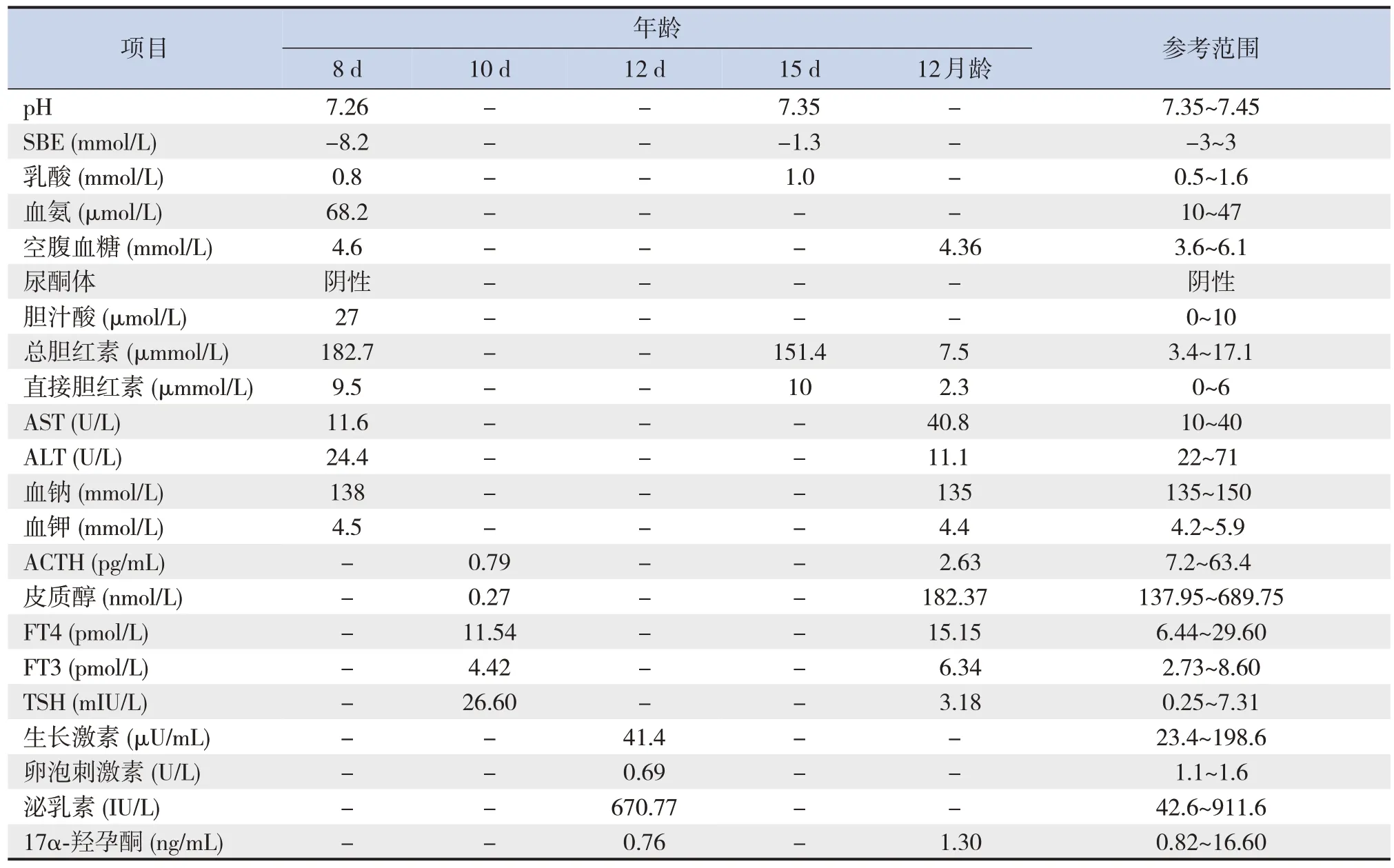
表1 患兒實驗室檢查結果
3 多學科診療
3.1 新生兒重癥監護病房初診
該患兒生后第1天因一過性低血糖及呼吸暫停入院,經治療后病情趨于穩定。但生后第8天心臟彩超提示左室射血分數降低,同時出現氣促及代謝性酸中毒表現,診斷為急性心功能不全。立即予以吸氧、強心治療,并積極完善相關檢查尋找潛在病因。實驗室檢查示ACTH和皮質醇水平顯著降低,考慮患兒存在腎上腺皮質功能不全,推測腎上腺皮質功能不全可能是導致患兒心功能不全的主要原因。
3.2 心內科會診
多種病因可導致新生兒急性心功能不全,主要可分為結構性心臟病和心肌病2大類(圖1)。其中結構性心臟病是新生兒心功能不全的最常見病因。該患兒心臟彩超和心臟增強CT除提示PDA外,未提示其他心臟結構異常。患兒生后早期喂養建立順利、早期撤離有創機械通氣,體格檢查示心前區未聞及雜音,不符合血流動力學不穩定PDA,因此不支持由PDA導致的心功能不全。進一步則需考慮是否存在圍產期疾病、心律失常、心肌病或全身性疾病引起患兒急性心功能不全。本例患兒皮質醇水平顯著降低,皮質醇在維持血管張力和腎上腺素能受體對兒茶酚胺的反應起著非常關鍵的作用,皮質醇水平不足會導致心率、心肌收縮力下降和血壓下降,最終導致心功能不全。本例患兒顯著降低的皮質醇水平可能是其急性心功能不全的主要原因。因此治療上主要為積極治療原發病,其次可予以強心、利尿、限液等支持治療。
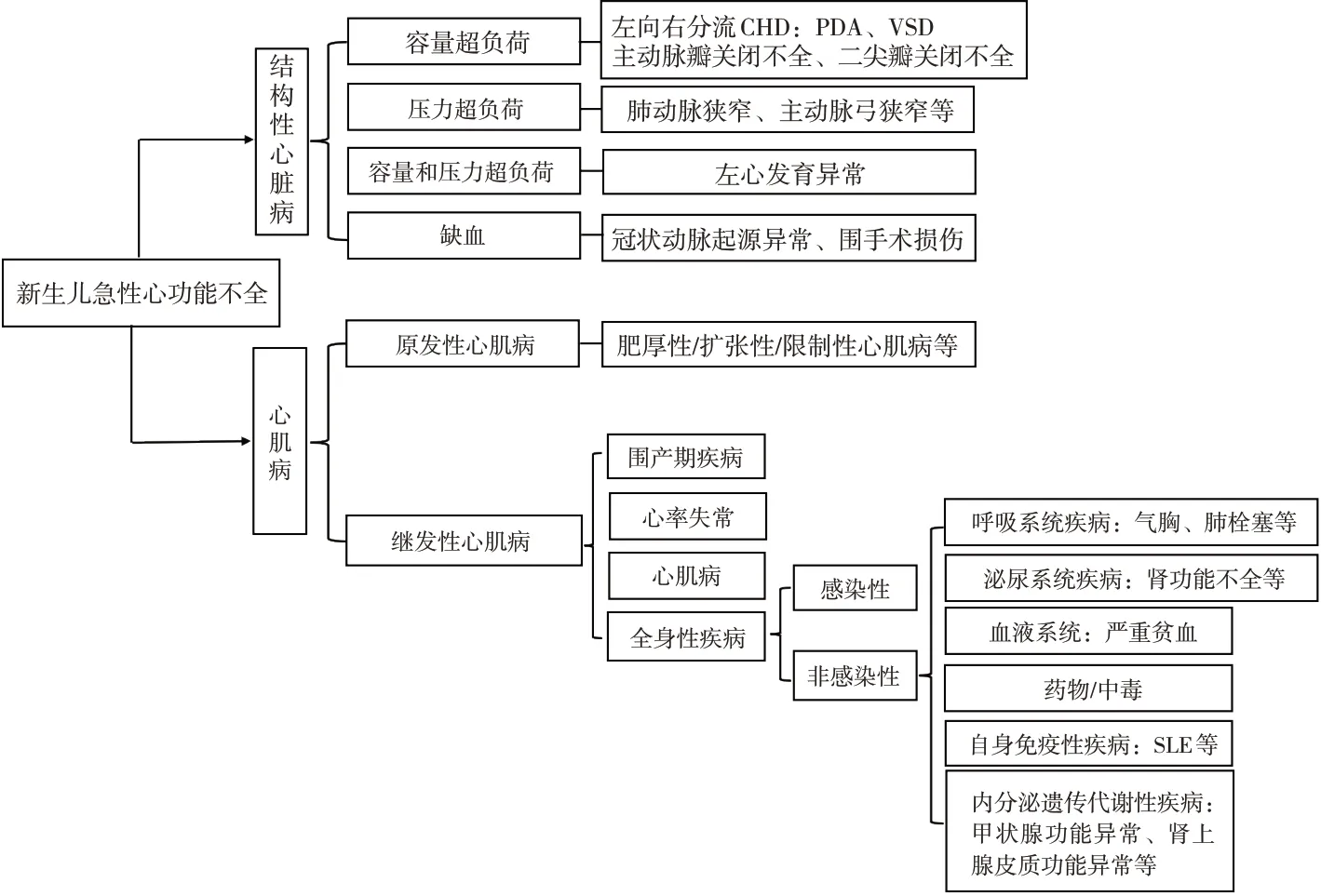
圖1 新生兒急性心功能不全的病因鑒別診斷 [CHD]先天性心臟病;[PDA]動脈導管未閉;[VSD]室間隔缺損;[SLE]系統性紅斑狼瘡。
3.3 內分泌遺傳代謝科會診
患兒以急性心功能不全起病,實驗室檢查提示ACTH和皮質醇水平顯著降低,考慮診斷腎上腺皮質功能不全。患兒出生史及家族史無特殊;生長發育良好,面容無特殊,皮膚無色素沉著;無電解質紊亂、垂體其他激素水平基本正常且垂體磁共振成像無異常,考慮是孤立性ACTH缺乏,新生兒期孤立性ACTH缺乏多是涉及垂體發育的基因突變或基因表達調控異常導致的,建議完善相關基因檢測。最終患兒基因檢測出TBX19基因存在復合雜合突變,分別來自父母,因此明確診斷TBX19基因變異所致CIAD。CIAD是一種罕見的常染色體隱性遺傳病,是由于ACTH缺乏導致繼發性腎上腺皮質功能不全,而垂體前葉其他激素水平正常。CIAD 臨床分型包括早發型完全性、部分性和晚發型。早發型完全性CIAD 的ACTH 和皮質醇水平更低,均會在新生兒期出現臨床表現[6]。CIAD 典型的臨床表現為頑固性低血糖、膽汁淤積和驚厥。少數CIAD 病例存在心肌病、Arnold-Chiari Ⅰ型畸形、47,XXX 綜合征、單側后鼻道閉鎖[6]。目前以心功能不全為主要表現的新生兒期起病的CIAD,暫未見報道。皮質醇水平可以通過多種機制作用于心血管系統,低水平的皮質醇可能是CIAD導致急性心功能不全的潛在機制。關于新生兒期起病的CIAD治療文獻報道少見。現有文獻中CIAD治療基本均為氫化可的松替代治療,但氫化可的松替代治療劑量尚無統一推薦[7-8]。CIAD如未早期識別,可導致新生兒期20%的病死率[9],及時的激素替代治療有效降低反復低血糖和難治性驚厥的風險,顯著改善預后。已有文獻報道中大部分新生兒期起病的CIAD經早期氫化可的松替代治療,預后良好[9-10]。該患兒存在急性心功能不全,病情危重,因此急性期可考慮使用較大劑量[100 mg/(m2·d)]氫化可的松沖擊治療3 d,以盡快控制代謝紊亂,臨床癥狀好轉后,盡快減少氫化可的松劑量至維持量[20 mg/(m2·d)]。
3.4 分子診斷中心會診
TBX19基因變異是早發型完全性CIAD 的最主要原因。垂體分泌的ACTH是在下丘腦促腎上腺皮質激素釋放激素作用下,刺激垂體前葉首先合成阿黑皮素原(proopiomelanocortin, POMC),經蛋白質轉換酶裂解后生成ACTH。轉錄TBX19基因對垂體前葉POMC細胞的發育分化至關重要。TBX19基因屬于T-box基因家族,TBX19基因可通過T-box基因結構域與POMC啟動子結合,激活POMC基因轉錄,誘導垂體分化表達POMC的細胞系,變異后POMC基因轉錄減少,影響ACTH產生。該患兒基因檢測顯示TBX19基因存在復合雜合變異,該變異在HGMD數據庫中尚無相關文獻報道。通過對患兒父母進行Sanger測序,父親攜帶c.917-2A>G,母親攜帶c.608C>T,判定為可疑致病變異[11],結合臨床表型考慮該2個變異為該患兒的致病原因。
3.5 新生兒重癥監護病房診斷思路總結
該患兒急性起病,以心功能不全為主要特征。實驗室檢查提示ACTH和皮質醇水平顯著降低,但垂體磁共振成像正常,基因檢測示TBX19基因復合雜合變異,分別來自父母,明確診斷TBX19基因變異所致CIAD,早期予以氫化可的松治療。以心功能不全為主要表現的CIAD目前未見報道。該患兒臨床僅表現為生后早期一過性低血糖及急性心功能不全,住院期間血糖、轉氨酶、膽汁酸及直接膽紅素水平基本正常,未出現頑固性低血糖、驚厥或膽汁淤積,這可能與早期診斷及早期氫化可的松替代治療有關。由于該患兒生后早期僅出現過一次低血糖,因此在該病例診治過程中,極易忽視低血糖的線索。
4 治療經過及轉歸
患兒入院后胸部X線片示左側少量氣胸,繼續予常頻機械輔助通氣、靜脈補液及抗感染治療。生后第2天成功撤離呼吸機,病情趨于穩定。生后第8 天復查心臟超聲,結果提示左室射血分數55%,PDA(2.6 mm),二尖瓣、三尖瓣中重度反流。之后24 h 內出現氣促,血氣分析提示代謝性酸中毒。立即予以鼻導管吸氧、糾酸及強心治療,患兒氣促緩解不明顯,隨訪心臟超聲提示左室射血分數進行性降低。生后第12 天診斷腎上腺皮質功能不全后予氫化可的松治療,后臨床癥狀迅速好轉。生后第21 天基因診斷結果示TBX19基因的復合雜合突變,突變分別來自父親和母親,患兒明確診斷TBX19基因變異所致CIAD。生后第41天患兒順利出院。患兒住院期間診療經過詳見圖2。出院后繼續口服氫化可的松治療,目前患兒18 月齡,居家監測血糖穩定;無驚厥、膽汁淤積及心功能不全等表現,生長發育良好,12 月齡時門診隨訪甲狀腺激素及17α-羥孕酮基本正常。

圖2 診療經過 [EF]射血分數;[ACTH]促腎上腺皮質激素;[MRI]磁共振成像;[PDA]動脈導管未閉。
5 小結
該患兒住院期間突發不明原因急性心功能不全。在病因尋找方面通過多學科診療,最后通過激素水平測定、垂體磁共振成像及基因結果,明確診斷TBX19基因突變所致CIAD。早期予以氫化可的松替代治療,預后良好。以下是我們在本病例診治過程中的幾點體會:(1)新生兒急性心功能不全需警惕內分泌遺傳代謝性疾病,應盡早行皮質醇檢測。(2)CIAD 如未早期識別,可導致新生兒死亡,針對該疾病,早期診斷及治療對預后至關重要。(3)CIAD典型臨床表現為反復低血糖、膽汁淤積及驚厥,基因檢測是確診依據。部分患兒可無上述典型表現,因此在臨床危重癥新生兒救治過程中,需警惕該疾病,建議盡早行基因檢測。
作者貢獻聲明:李淑涓負責撰寫文章、病例采集與分析;胡黎園、張蓉、楊琳、奚立、劉芳、曹云、周文浩負責病例分析指導;程國強負責文章指導與修改。
利益沖突聲明:所有作者聲明無利益沖突。
猜你喜歡
中華養生保健(2020年8期)2021-01-14 01:13:58
家庭醫學(下半月)(2019年9期)2019-10-12 08:04:06
家庭醫學(下半月)(2019年8期)2019-09-25 09:02:00
護士進修雜志(2017年2期)2017-02-26 14:32:29
媽媽寶寶(2017年3期)2017-02-21 01:22:12
罕少疾病雜志(2016年4期)2016-03-11 16:34:39
中國衛生標準管理(2015年18期)2016-01-20 09:27:12
中國衛生標準管理(2015年13期)2016-01-15 02:58:25
中國醫藥科學(2015年2期)2015-02-27 12:32:09
中國衛生標準管理(2015年8期)2015-01-26 18:08:35
