餅干中紫草紅色素檢測方法的建立
2024-05-26 09:30:26邊文文
農產品加工 2024年7期
王 彬,何 沖,安 瑜,邊文文
(陜西省產品質量監督檢驗研究院,陜西 西安 710048)
食品添加劑因具備增加食品風味、改善食品品質、提高食品營養價值等功能,被譽為現代食品工業魔法師[1]。紫草紅是從紫草科紫草屬植物根中提取的紅色素,主要成分是紫草素及其衍生物,為暗紫紅色結晶品、紫紅色黏稠膏狀或紫紅色粉末,溶于有機溶劑和油脂,不溶于水,但溶于堿液[2-4];可作為食品添加劑應用于飲料、餅干、果酒、肉制品、點心等食品中,天然健康,還具有清熱消毒、消炎抗菌、防病抗癌等藥理性功效[5-7]。紫草紅色素因具性能穩定、色澤艷麗、吸附著色力強、持久力久等優點,在食品、保健品、藥品、生物制品等領域有著廣闊的應用前景。
在GB 2760—2014《食品安全國家標準食品添加劑使用標準》[8]中明確規定果汁飲料類、糕點、餅干、雪糕、果酒等食品中可添加的紫草紅色素最大限量為0.1 g/kg。目前,食品色素產業發展迅猛,但關于檢測紫草紅色素的方法大多用于藥品,而食品中檢測方法尚未完善,檢測方法滯后給食品安全帶來了巨大的隱患[9]。梁占麗等人[10]建立了軌道阱高分辨質譜法,對飲料中紫草紅進行了分析測定,檢出限為200~500 μg/kg,相對標準偏差為4.0%~9.2%,可以簡單快速地對飲料中紫草紅色素進行快速篩選和分析。以食品作為基質的檢測方法未見到相關的報道。通過對前處理方法進行摸索,優化色譜條件,建立高效液相色譜法測定餅干中紫草紅色素的含量,為食品中紫草紅色素安全監控提供技術支撐。
1 材料與方法
1.1 材料與試劑
紫草紅標準品(20 mg),上海安譜實驗科技股份有限公司提供;乙腈(色譜純)、甲醇(色譜純),賽默飛世爾科技有限公司提供;石油醚(分析純),天津市富宇精細化工公司提供;無水乙醇(色譜純),美國Honeywell 公司提供;甲酸(優級純),天津市科密歐化學試劑公司提供;纖麩木糖醇消化餅干,東莞思朗食品有限公司提供。
1.2 儀器與設備
Waters E2695 型高效液相色譜儀,美國Waters公司產品;ST-16R 型高速離心機,美國ThermoFisher 公司產品;ZORBAX Eclipse XDB-C18柱(4.6 mm×250 mm,5 μm) 色譜柱,美國Agilent 公司產品;AUTO 型全自動氮吹儀,廈門瑞科儀器有限公司產品;KQ-250DE 型超聲波清洗器,昆山市超聲儀器有限公司產品;XS-20S 型電子分析天平,瑞士METTLER-TOLEDO 公司產品。
1.3 試驗方法
1.3.1 紫草紅標準溶液的配制
稱取0.01 g 紫草紅標品(精確至0.000 1 g),置于100 mL 容量瓶,用甲醇溶解,并定容,配制成質量濃度為100 μg/mL 紫草紅標準溶液,于-18 ℃條件下避光保存。
1.3.2 樣品前處理
稱取餅干5 g(精確至0.01 g),加入20 mL 甲醇提取液,旋渦混勻,超聲20 min,經過5 g 無水硫酸鈉脫水后,以轉速5 000 r/min 離心5 min,取上清于40 ℃條件下氮吹,甲醇- 0.1%甲酸= 85∶15(V/V) 流動相定容至1 mL,經0.22 μm PTFE 濾頭過濾待測。
1.3.3 色譜條件
色譜柱采用Agilent ZORBAX Eclipse XDB-C18柱(4.6 mm×250 mm,5 μm);流動相A 為甲醇,B 為0.1%甲酸(V/V=85∶15),流速1.0 mL/min,檢測波長516 nm,進樣量10 μL,柱溫35 ℃,采用等梯度洗脫。
1.3.4 標準曲線的測定
依次取適量標準溶液,稀釋為0.5,1,2,5,10,50,100 μg/mL 7 個質量濃度梯度,以紫草紅質量濃度為橫坐標,峰面積平均值為縱坐標繪制紫草紅標準曲線。
1.3.5 樣品加標回收率測定
按上述前處理條件進行加標回收試驗,在餅干中分別添加質量濃度為10.0,50.0,200.0 mg/kg 的紫草紅標準溶液,計算回收率和相對標準偏差(RSD)。
2 結果與分析
2.1 提取試劑的選擇
食品中的紫草紅常與其他人工合成色素混合使用,因此要去除人工合成色素的干擾。食用色素易溶于水,因此為了避免提取到食用色素,選用有機試劑進行提取。由圖1 可知,以1.0 mg/kg 紫草紅加標時,石油醚和無水乙醇作為提取劑時回收率較低,僅為31.25%和67.45%,乙腈回收率為81.21%,甲醇作為提取溶劑,相較乙腈提取回收率要更高,為100.32%。可見甲醇提取效果較好,雖帶有一定的雜質,但對紫草紅出峰情況影響不大,幾乎不受人工合成色素的影響;且基線噪音小,峰形較美觀,因此選擇甲醇作為提取劑。

圖1 不同提取劑對應的紫草紅色素色譜圖
不同提取劑對應的紫草紅色素色譜圖見圖1,以餅干為基質4 種提取溶劑回收率比較見表1。
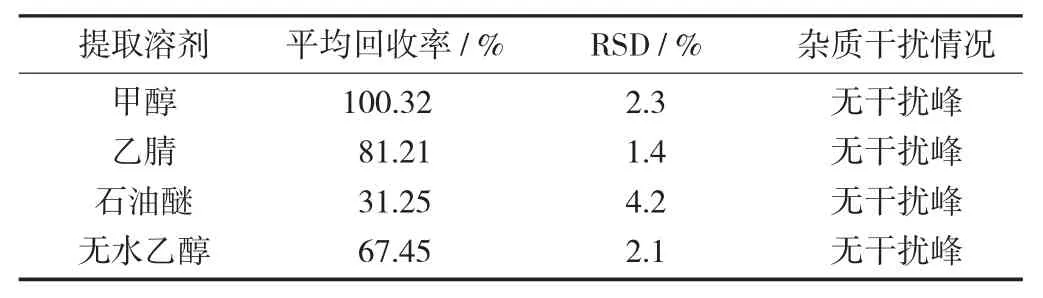
表1 以餅干為基質4 種提取溶劑回收率比較
由表1 可知,以甲醇提取劑時,噪音干擾小,基線平穩且回收率高,相對標準偏差在合適范圍內。
2.2 提取方式選擇
餅干通常含有巧克力、可可脂、乳化劑等物質,制樣后添加物多的餅干不容易分散,直接浸提法提取效果不佳。試驗對比超聲不同時間的提取效果,選取時間分別為10,20,30,40 min。超聲提取達到20 min 后,延長時間不能提高回收率。因此,確定了直接超聲浸提為最佳紫草紅色素提取方法,提取溫度要嚴格控制在60 ℃以下,溫度過高容易導致紫草素分解。
2.3 流動相及比例的選擇
紫草素一般選擇甲醇-緩沖鹽體系和乙腈-甲酸作為流動相[11-12],緩沖鹽使用不當時易析出,因此試驗測試乙腈-水和甲醇-水作為流動相時,紫草素峰形容易出現拖尾,峰較寬。流動相pH 值降低后,紫草素峰拖尾得到改善。
不同流動相下紫草紅標準色譜圖(50 μg/mL)見圖2。

圖2 不同流動相下紫草紅標準色譜圖(50 μg/mL)
由圖2 可知,紫草紅色素以乙腈-水為流動相時出峰時間較早且有些許拖尾現象,甲醇-0.1%甲酸為流動相時,峰不拖尾且對稱;使用甲醇-0.1%甲酸時,優化比例后,紫草紅出峰時間為5.273 min,峰形較好,可縮短分析時間,同時甲醇提取出來的雜質出峰較早,可與紫草紅實現分離。
2.4 檢測波長的選擇
紫草紅色素光譜圖見圖3。

圖3 紫草紅色素光譜圖
由圖3 可知,紫草素在217 nm 和516 nm 均有吸收,波長217 nm 下容易受其他雜質干擾,而516 nm下色譜圖基線平整,干擾雜質相對較少且目標峰附近無雜質干擾峰,可較好地簡化凈化過程,因此確定檢測波長為516 nm。
2.5 方法線性范圍及檢出限
紫草紅色素的標準曲線見圖4。
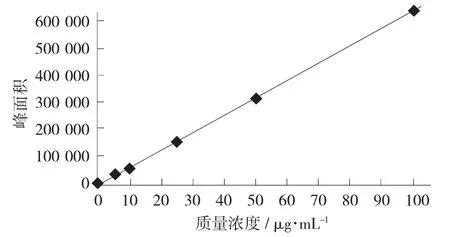
圖4 紫草紅色素的標準曲線
由圖4 可知,質量濃度0.5~100.0 μg/mL 時表現出良好的線性關系。通過樣品基線噪音在紫草紅標準工作溶液的響應信號進行比較,以3 倍信噪比(S/N=3) 所對應樣品中的紫草紅質量濃度為檢出限進行計算,得到方法檢出限為0.2 mg/kg。
紫草紅色素的線性范圍、線性方程、相關系數、檢出限見表2。

表2 紫草紅色素的線性范圍、線性方程、相關系數、檢出限
2.6 回收率和精密度試驗
加標回收試驗結果表明,紫草紅色素的平均回收率為88.1%~96.7%,相對標準偏差(RSD) 為2.7%~5.8%,表明該方法精確度高、準確性好,能夠較準確地對餅干中紫草紅色素進行定量測定。
餅干中紫草紅色素回收率和精密度試驗結果(n=6) 見表3。
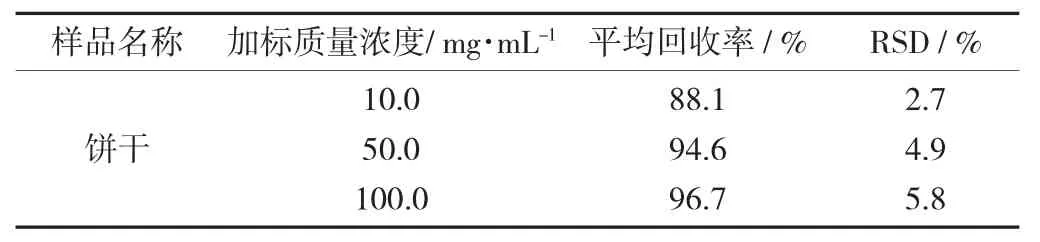
表3 餅干中紫草紅色素回收率和精密度試驗結果(n=6)
2.7 實際餅干樣品檢測
采用該檢測方法對市面上可能含有紫草紅色素餅干進行檢測,其中夾心餅干測出紫草紅,威化餅干未檢測出紫草紅色素。相對標準偏差為2.2%~3.2%,結果表明該方法的精密度與重復性較好。
不同餅干樣品中紫草紅色素的測定結果(n=6)見表4。
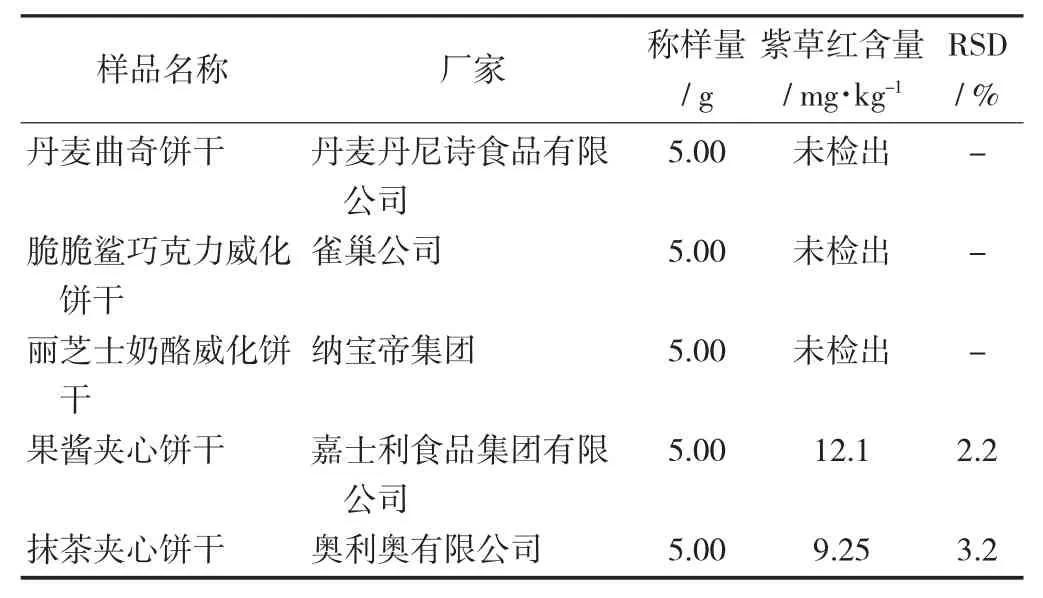
表4 不同餅干樣品中紫草紅色素的測定結果(n=6)
3 結論
基于紫草紅色素檢測方法的滯后,建立了一種測定餅干中紫草紅色素含量的高效液相色譜法。餅干樣品經甲醇超聲提取后定容上機,采用Agilent ZORBAX Eclipse XDB-C18色譜柱,以甲醇- 0.1%甲酸=85∶15(V/V) 為流動相,檢測波長為516 nm,柱溫為35 ℃,采用等度洗脫。結果表明,在該色譜條件下紫草紅色素質量濃度為0.5~100.0 μg/mL 時呈良好線性關系,相關系數大于0.999 9,方法檢出限為0.2 mg/kg,平均加標回收率為88.1%~96.7%,相對標準偏差(n=6) 為2.7%~5.8%。表明該方法精確度高、準確性好,能夠較準確地對餅干中紫草紅色素進行定量測定。用此方法對5 種市銷售餅干中紫草紅色素含量進行測定,相對標準偏差為0.8%~3.2%,證實其具有前處理方法操作簡單、分析時間短、加標回收率高、檢測波長高、抗干擾能力強和檢出限低的特點,可適用于餅干中的紫草紅色素含量的檢測。此方法的建立填補了紫草紅在餅干食品中檢測方法的空缺,為食品中紫草紅色素的定量檢測提供了理論參考。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
