基于全基因組重測序技術對平菇白色變異菌株的鑒定及遺傳變異研究
2024-11-07 00:00:00夏會楠郝劉斌田雨李海康王春霞鄭素月
江蘇農業科學 2024年18期


摘要:以生產上收集的一個平菇白色變異菌株(Po50)、黑色出發菌株(Po51)和一個白色生產菌株(Po9)為材料,采用拮抗、酯酶同工酶和全基因組重測序方法對3個菌株進行鑒定。拮抗和酯酶同工酶結果表明,白色變異菌株和黑色出發菌株之間無明顯拮抗和酶譜差異,與生產平菇白色菌株差異較大;進一步通過全基因組重測序技術鑒定結果表明,3個菌株鑒定出大量的SNP(單核苷酸多態性位點)、InDel(插入缺失位點)、SV(結構變異位點)。平菇白色變異菌株Po50檢測到SNP、InDel突變總數為1 504 391個,生產菌株Po9檢測到SNP、InDel突變總數為1 371 818個,黑色出發菌株Po51檢測到SNP、InDel突變總數為1 501 877個,通過生物信息手段分析白色變異菌株黑色出發菌株以及對照菌株Po9基因組間的結構差異,獲得遺傳變異圖譜,可以對變異菌株進行快速精準鑒定。
關鍵詞:平菇;變異菌株;全基因組重測序技術;酯酶同工酶
中圖分類號:S646.1+40.1 文獻標志碼:A
文章編號:1002-1302(2024)18-0041-09
收稿日期:2023-09-20
基金項目:河北省現代農業產業技術體系食用菌創新團隊建設專項資金(編號:HBCT2023090207);河北省農業科技成果轉化項目(編號:202360101010014)。
作者簡介:夏會楠(1998—),女,河北承德人,碩士,從事食用和藥用真菌研究。E-mail:2199797807@qq.com。
通信作者:鄭素月,博士,教授,碩士生導師,主要從事食用菌遺傳育種研究。E-mail:zhengsuyue@sina.com。
平菇(Pleurotus ostreatus)栽培廣泛,含有人體所需的大量生物活性物質、礦物質和維生素,具有很高的營養價值。由于其栽培技術簡單、生長周期短、收益快、適應性強,為食用菌生產主栽品種之一。平菇產業中存在的主要問題是育種工作落后,品種單一,生產缺乏優良品種,因此平菇新品種種質資源選育十分重要,經常規鑒定后在分子層面進行精準分析,有利于更方便快捷地選育新品種。聶興華等以野生板栗為試驗材料,通過重測序技術確定46份野生板栗的分類,發現野生板栗和栽培板栗屬于同種,為我國野生板栗提供了更準確的邊界范圍[1]。沈秀芬等通過對5個香菇進行重測序,分析這些菌株中的插入/缺失位點開發InDel標記,可將44個香菇菌株分為4個亞群[2]。侯炳豪等以鐵觀音茶樹為試驗材料,通過重測序技術對鐵觀音無性繁殖后代進行遺傳差異研究,根據重測序數據得出在全基因組和編碼區范圍內,各個樣本之間總變異位點數均差別不大,各樣本中涉及突變的基因數基本一致,推測鐵觀音品種的種性在無性繁殖過程中總體保持穩定[3]。
全基因組重測序技術對已知基因序列的物種進行不同個體間的基因組重測序,并在此基礎上對個體進行差異性分析[4],通過序列對比,在平菇基因組水平上開發大量分子標記如鑒定食用菌中大量的SNP單核苷酸多態性位點、InDel插入缺失位點、SV結構變異位點[5-6],通過生物信息手段,分析平菇白色變異菌株、栽培菌株Po9以及黑色出發菌株基因組間的結構差異,獲得遺傳變異圖譜。全基因組重測序技術有助于快速發現重要性狀的遺傳變異以及對變異菌株精準鑒定[7-8]。
1 材料與方法
1.1 試驗材料
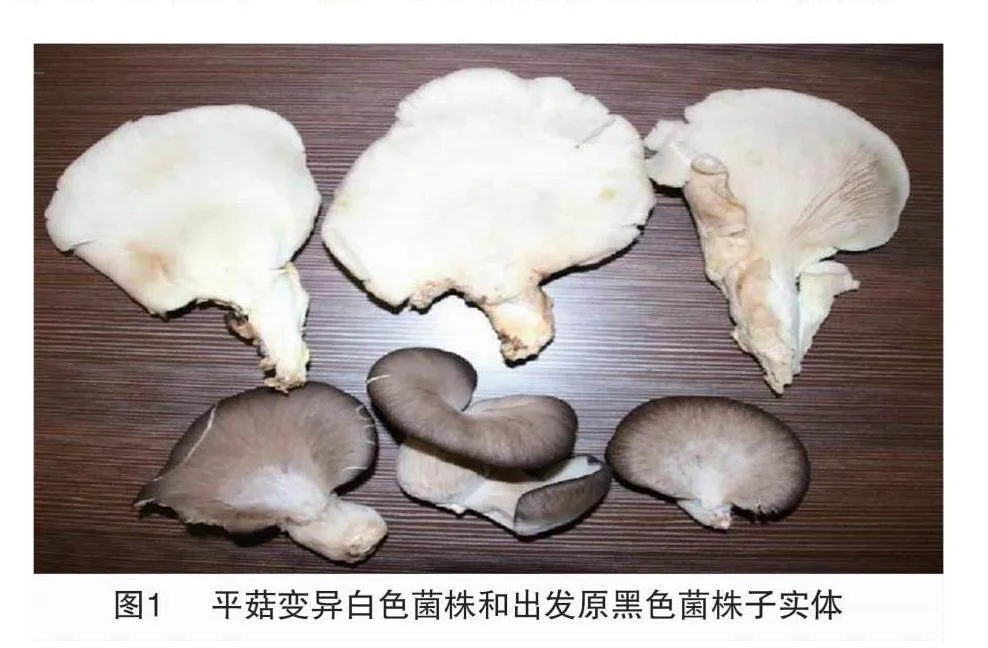
試驗于2023年2—5月在河北工程大學食用菌研究室內進行,在平菇系統選育過程中,發現變異白色平菇菌株1個,變異的白色子實體和對應的出發菌株形態見圖1。對采集的白色變異菌株子實體和黑色出發菌株子實體進行組織分離,獲得純菌種,白色變異菌株編號為Po50,出發黑色菌株編號為Po51。生產上栽培的白色平菇編號為Po9,以上材料均由河北工程大學食用菌實驗室分離保藏。
1.2 試驗方法
1.2.1 拮抗和酯酶同工酶測定 以平菇89、平菇99、雙抗黑平以及生長上栽培的白平菇Po9作為對照菌株,對白色變異菌株和黑色出發菌株進行拮抗和酯酶同工酶測定[9]。
1.2.2 全基因組重測序技術
對平菇白色變異菌株Po50、黑色出發菌株Po51和生產對照白色菌株Po9進一步進行基因組重測序技術,文庫構建和重測序數據均由北京諾禾致源科技股份有限公司提供。
1.2.3 SNP/InDel檢測及注釋統計
采用GATK 4.0.9.0軟件進行MarkDuplicates、BaseRecalibrator處理后,再用GATK軟件中的HaplotypeCaller進行SNP、InDel檢測,并對變異位點進行質控標記。
2 結果與分析
2.1 白色變異菌株親緣關系鑒定
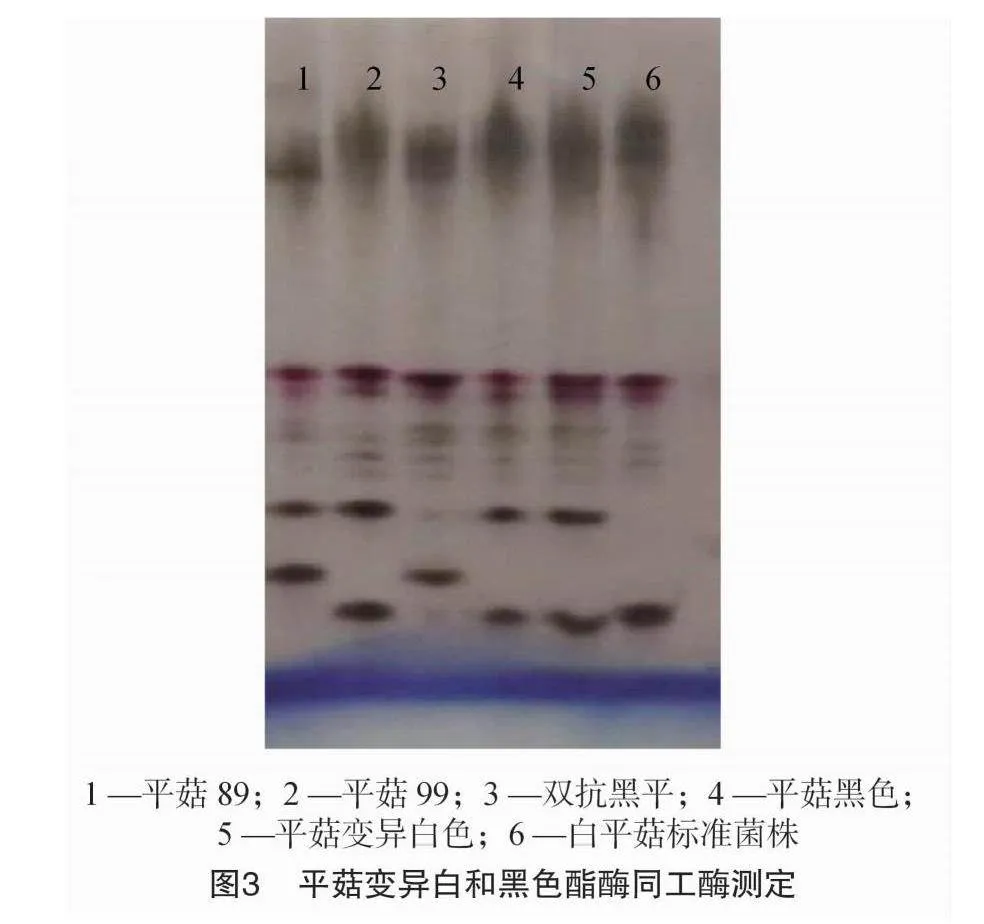
拮抗測定結果表明,平菇白色菌株Po50與出發黑色菌株Po51無拮抗作用,二者均與平菇99無拮抗作用(圖2)。酯酶同工酶圖譜顯示平菇白色變異菌株與原黑色菌株及標準99菌株酯酶同工酶圖譜相同,與2個標準菌株(平菇89、雙抗黑平、平菇黑色)和生產栽培白平菇Po9有較大差異(圖3),可以初步確定出發黑色菌株為平菇99的同物異名,而白色變異菌株在遺傳上與黑色出發菌株無顯著差異。
2.2 全基因組重測序技術
2.2.1 測序數據質量評估及對比
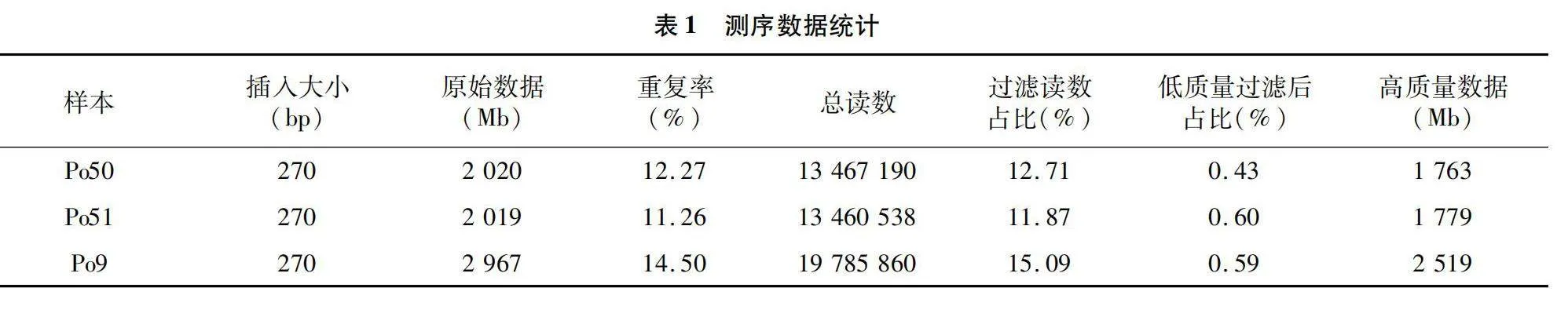
利用Illumina PE150測序,由于原始測序數據可能包含低質量序列、接頭序列等,為了保證信息分析結果的可靠性,需要過濾這些雜質,從而得到高質量clean data,共獲得2 020 Mb的平菇白色變異菌株、2 019 Mb的平菇黑色出發菌株、2 967 Mb的Po9基因組原始數據(raw data)。經質量評估和過濾雜質,最終平菇白色變異菌株獲得1 763 Mb高質量數據 clean data、平菇黑色出發菌株獲得1 779 Mb高質量數據clean data、Po9獲得2 519 Mb高質量數據clean data,采用測序平臺對樣品進行測序,數據統計見表1。
參考基因組對比結果將有效的測序數據通過BWA對比參考基因組得到最初對比結果,利用最初對比結果進行mapping率、duplication等的統計。重測序數據與參考數據樣品對比平菇白色變異菌株Po50檢測到的reads數目為11 754 734個,黑色平菇Po51共檢測到reads數目為11 862 522個,Po9菌株共檢測到16 799 756個 reads。整體對比率分別為80.48、81.11、75.83,覆蓋率分別為90.33%、90.32%、83.08%(表2)。
2.2.2 SNP及InDel突變檢測
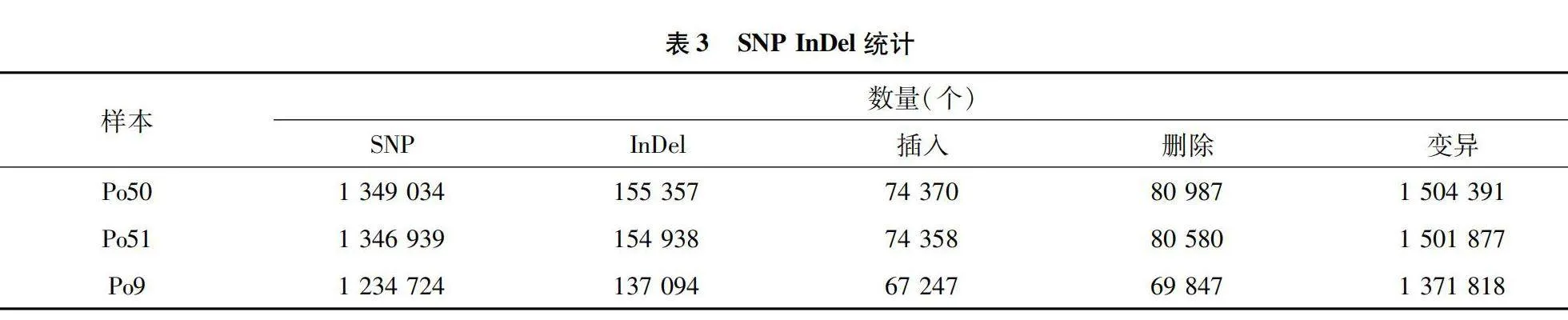
隨著食用菌基因組的公布,通過全基因組重測序以及生物技術挖掘平菇基因組SNP標記簡便快捷,SNP主要是指在基因組水平上由單個核苷酸的變異所引起的DNA序列多態性。SNP所表現的多態性只涉及到單個堿基的變異,這種變異可由單個堿基的轉換(transition)或顛換(transversion)所引起。將所測得的數據采用GATK軟件進行MarkDuplicates、BaseRecalibrator處理后,在利用GATK軟件中的HaplotypeCaller進行SNP檢測,最后在對檢測的結果進行質量過濾。最終得到SNP、InDel統計結果(表3),分別檢測到突變總數為1 504 391、1 501 877、1 371 818個。
2.2.3 SNP注釋
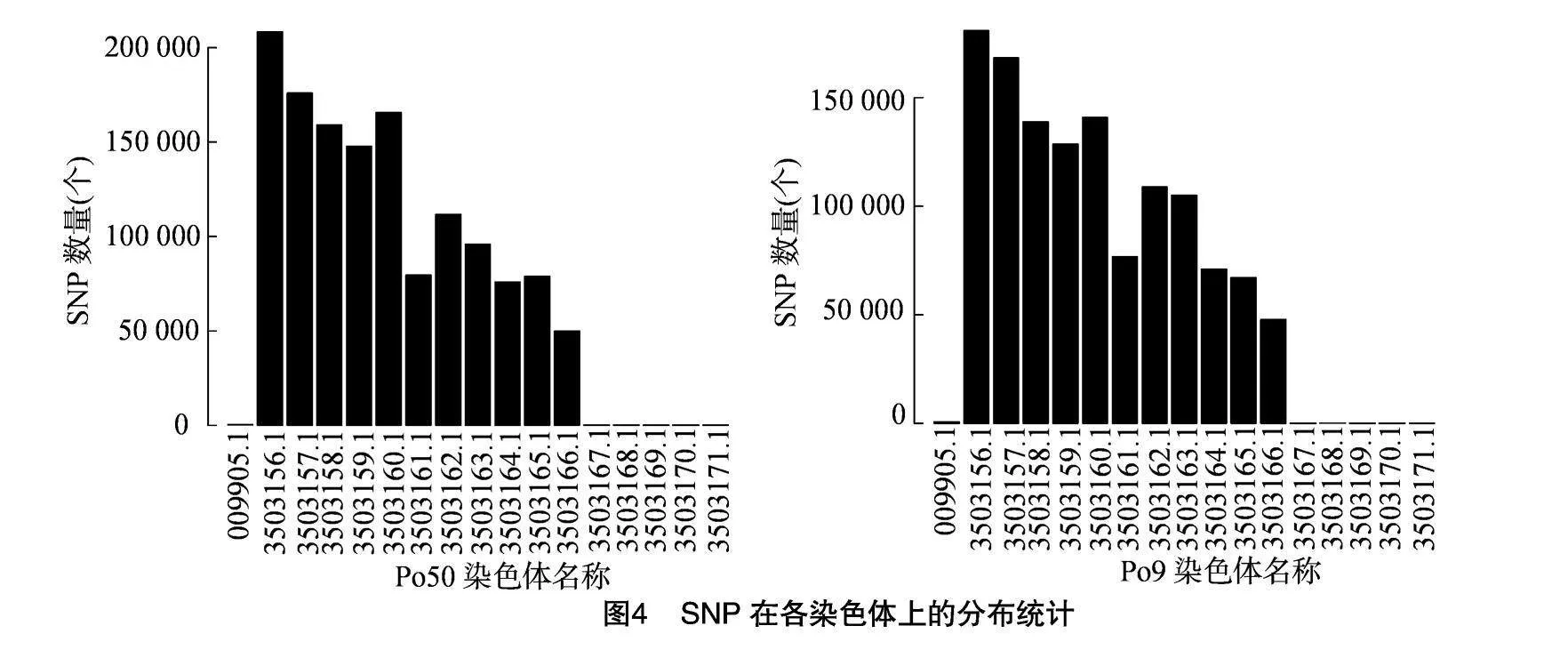
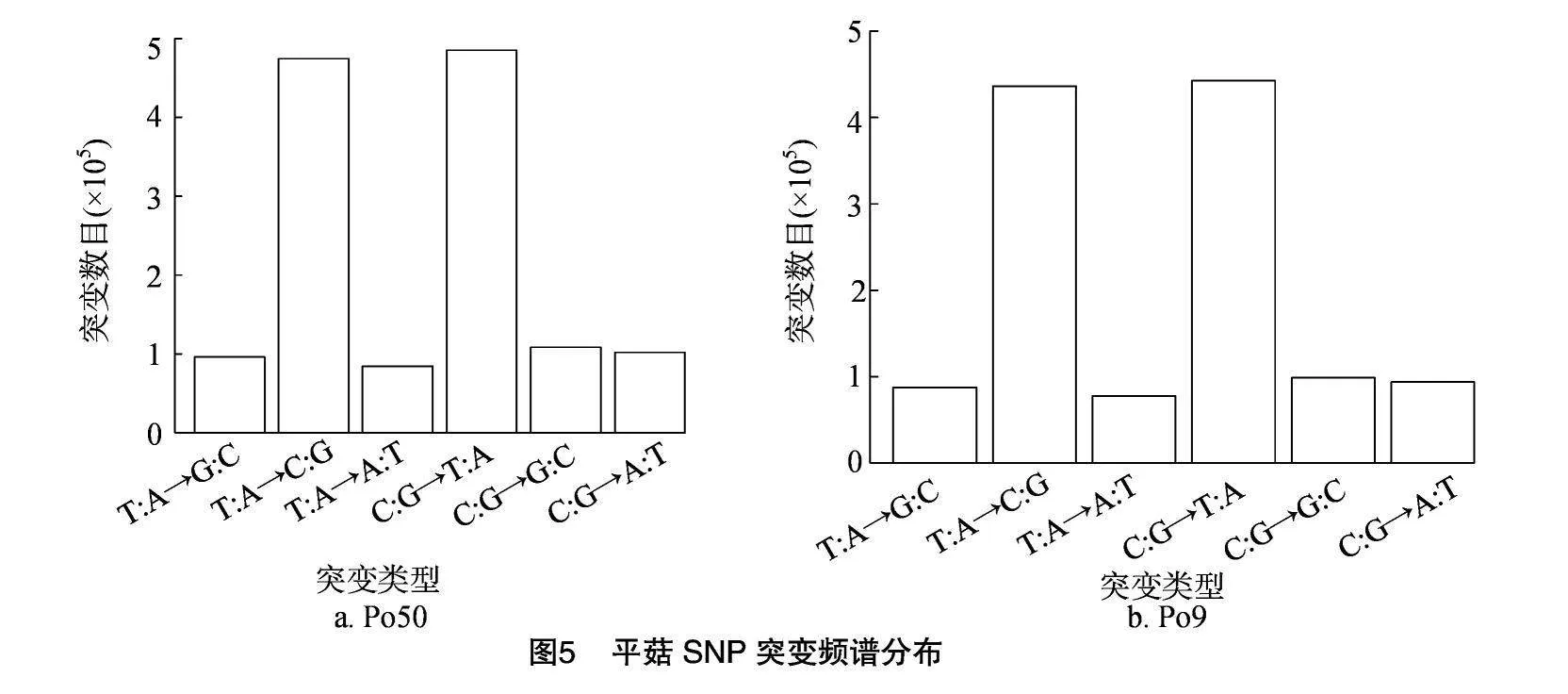
采用使用ANNOVAR對檢測到的SNP進行注釋,可對變異組合進行多層次的組合篩選。SNP在各染色體上分布統計見圖4,Po50在染色體3503156.1上SNP數目最多,其次是染色體3503157.1、3503160.1、3503158.1,在染色體3503167.1、3503168.1、3503169.1、3503170.1、3503171.1、009905.1上SNP數目最少。Po9在染色體3503156.1、3503157.1上 SNP數目最多,在染色體3503167.1、3503168.1、3503169.1、3503170.1、3503171.1、009905.1上SNP數目最少。通過突變頻譜分析可以直觀看出點突變包含6種類型:T∶A→G∶C,T∶A→C∶G,T∶A→A∶T,C∶G→T∶A,C∶G→G∶C 和 C∶G→A∶T,在Po51、Po50、Po9中各種突變類型的比例存在某種突變類型的偏好性,其中T∶A→C∶G、C∶G→A∶T類型的突變較多,突變頻譜分布見圖5。
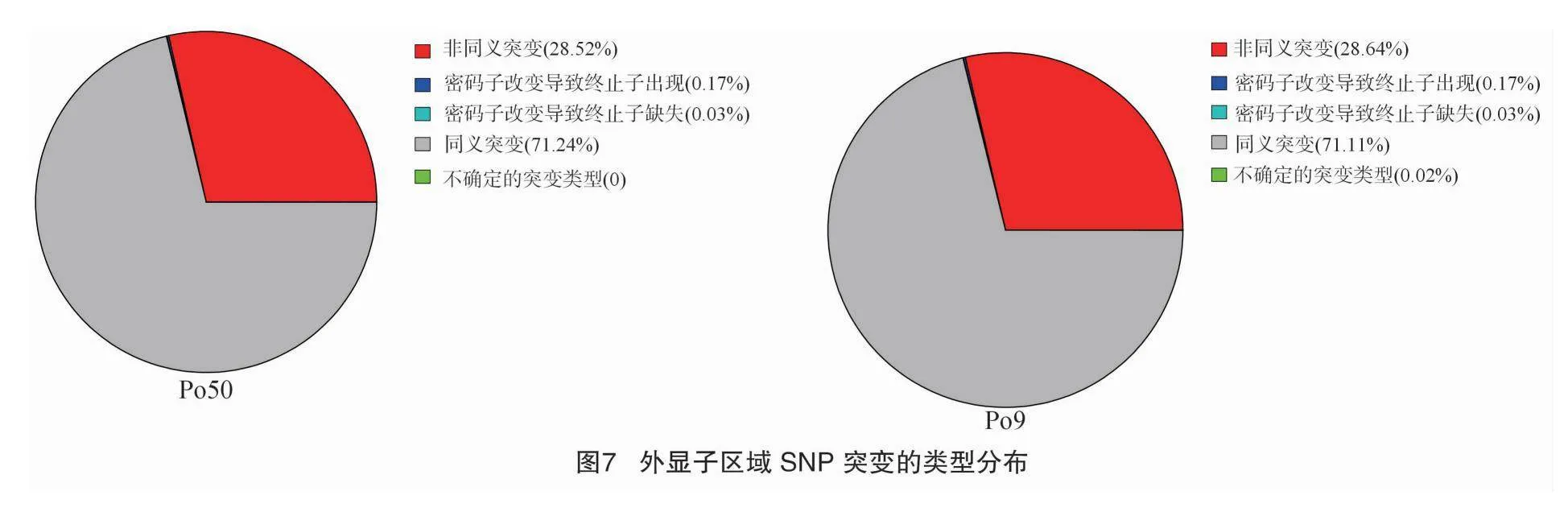
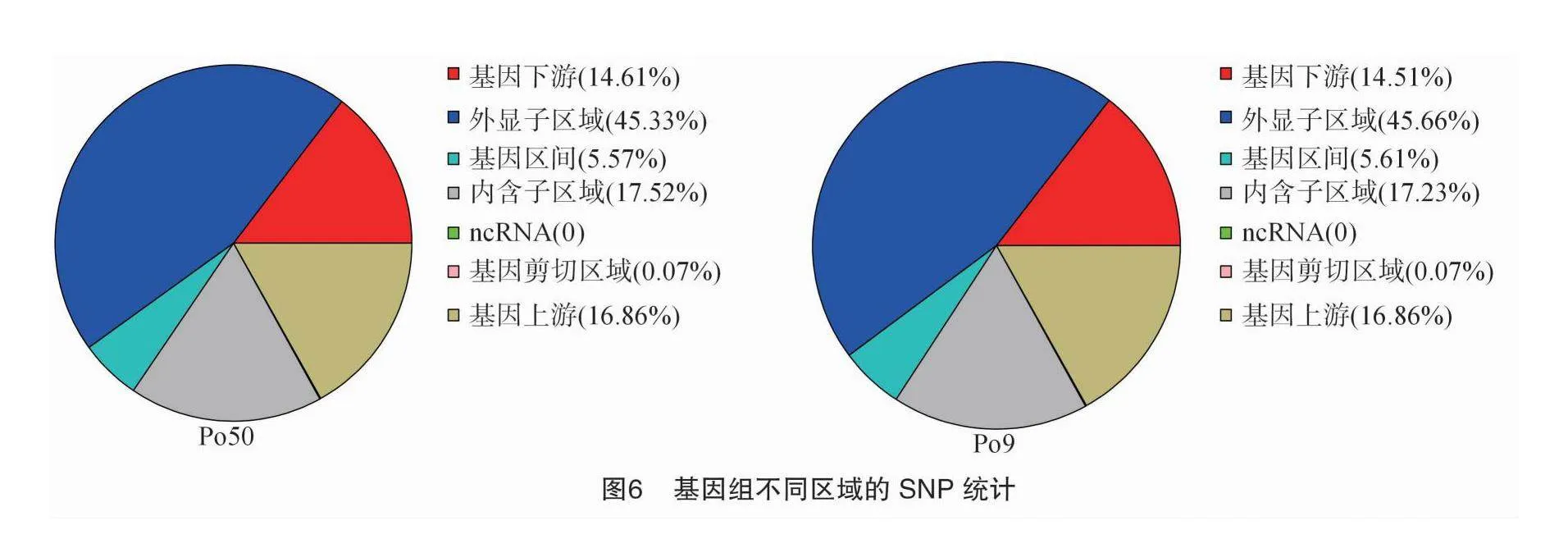
根據ANNOVAR的注釋結果,SNP在基因組各區域分布統計見圖6,SNP變異主要集中于發生在外顯子區域,比e71d63d306b1d50358201d0558687f6d4af4b42aab7079ad363ff0e68f703025例在45.33%~45.66%,發生在基因下游的SNP變異比例在14.51%~14.61%,基因上游、基因區間、內含子區域、ncRNA區域、基因剪切區域比例分別占16.86%、5.57%~5.61%、17.23%~17.52%、0、0.07%。其中,在外顯子區域發生同義突變的SNP比例為71.11%~71.24%,非同義突變為28.52%~28.64%,使基因轉錄提前終止突變為0.17%,失去終止密碼子突變為0.03%(圖7)。
2.2.4 InDel注釋及統計
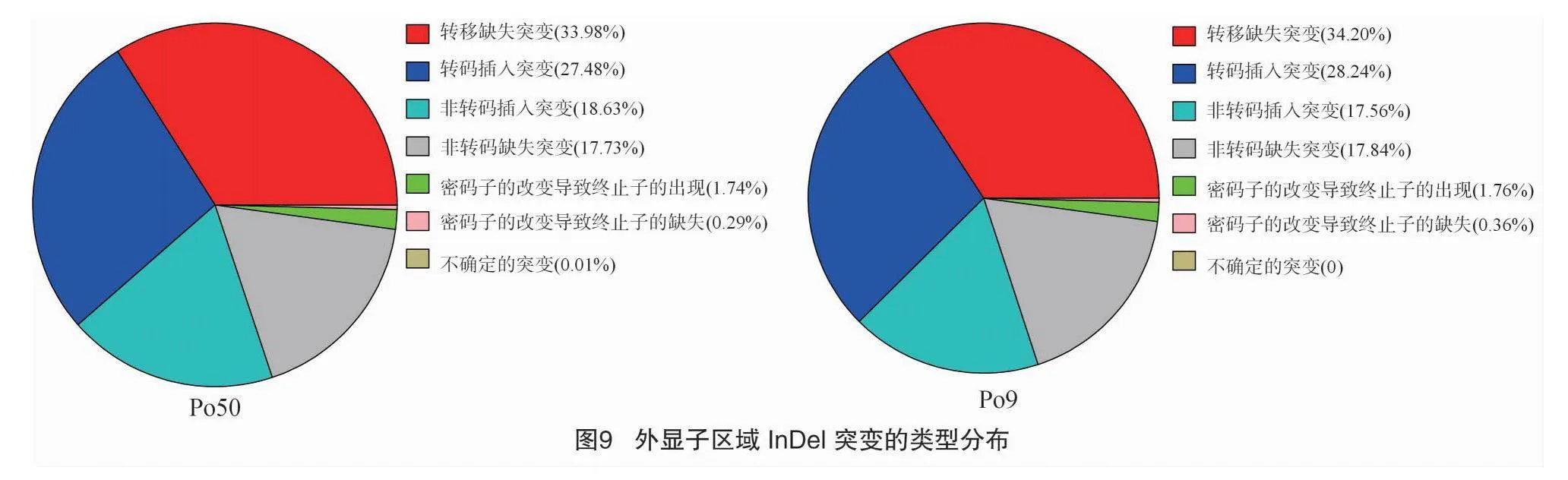
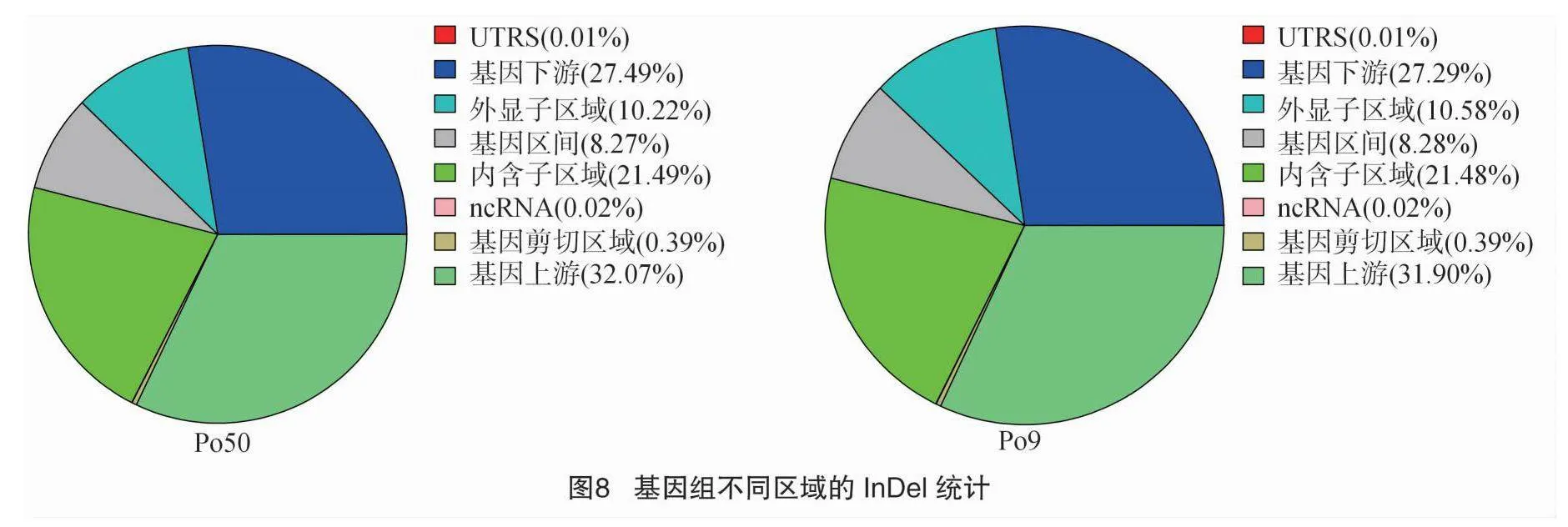
利用 ANNOVAR 對檢測出的 InDel 進行注釋,InDel突變檢測與注釋方法與SNP檢測注釋方法一致。 InDel在基因組各區域分布統計見圖8,InDel變異主要集中于發生在基因上游外顯子區域,比例在31.90%~32.07%;發生在基因下游的 InDel變異比例在27.29%~27.49%,基因區間、內含子區域、ncRNA區域、基因剪切區域比例分別占8.27%~8.28%、21.48%~21.49%、0.02%、0.39%。總體來說,InDel突變在
全基因組范圍內的表現與SNP突變基本一致。在外顯子區域內,移碼缺失突變所占比例最高,占33.98%~34.20%,移碼插入突變占外顯子區域突變總數的27.48%~28.24%,非移碼插入突變占17.56%~18.63%(圖9)。
在基因外顯子區域的SNP(InDel)位點中,部分位點導致基因編碼中氨基酸改變,而引起基因產物的突變,可能會影響平菇基因的生物學功能。
3 平菇差異基因的GO功能富集分析
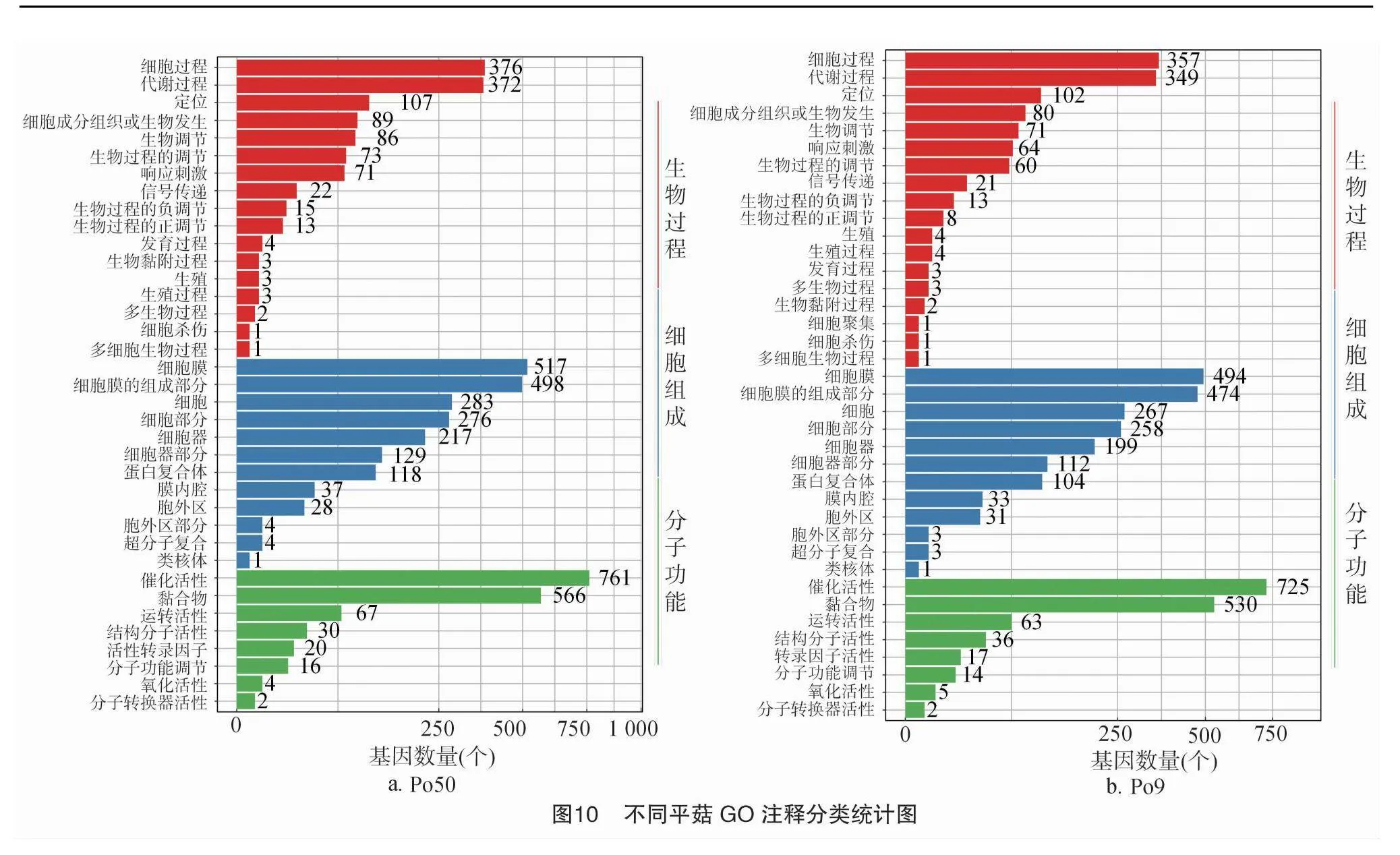
使用BLAST2GO v2.5對所測Po50、Po9基因進行GO功能注釋(圖10),Po50共注釋到5 760個基因,注釋到37個類別,主要包括生物過程(biological process,BP)、細胞組成(cellular component,CC)、分子功能(molecular function,MF)3個類別,其中CC數量最多,注釋到12個條目,總數為2 112個,最多的集中在細胞膜,占總數的24.5%,數量達到517個,其次是細胞和細胞部分(GO:0009696),分別占總數的13.4%和13.1%,數量為283個及276個;MF注釋8條,共有1 466個基因,最多的是催化活性部分(GO:0000155),占總數的51.9%,數量為761個,黏合物(GO:0046872)較少于活性催化部分,占總數的38.6%,數量為566個。其次是運轉活性、結構分子活性、活性轉錄因子,分別占總數的4.6%、2.0%、1.4%;氧化活性、分子轉換器活性最少,分別占總數的0.3%、0.1%。
Po9共注釋到5 386個基因,注釋到38個類別,主要包括生物過程、細胞組成、分子功能3個類別,其中CC數量最多,注釋到12個條目,總數為1 979個,占3個類別總數的43.8%。最多的集中在細胞膜(GO:0016021),占總數的25.0%,數量達到494個:細胞膜組成部分(GO:000021)少于細胞膜,占總數的24.0%,數量為474個,其次是細胞、細胞部分。MF注釋8條,共有1 392個基因,最多的是催化活性部分(GO:0000155),占總數的52.1%,數量為725個,黏合物(GO:0046872)較少于活性催化部分,占總數的38.1%,數量為530個。其次是轉錄活性,占總數的4.5%,數量為63個。在生物過程中Po9較Po50有1個基因注釋到細胞聚集類別中,生物過程及分子功能Po9涉及的基因數量,與Po50基本一致。
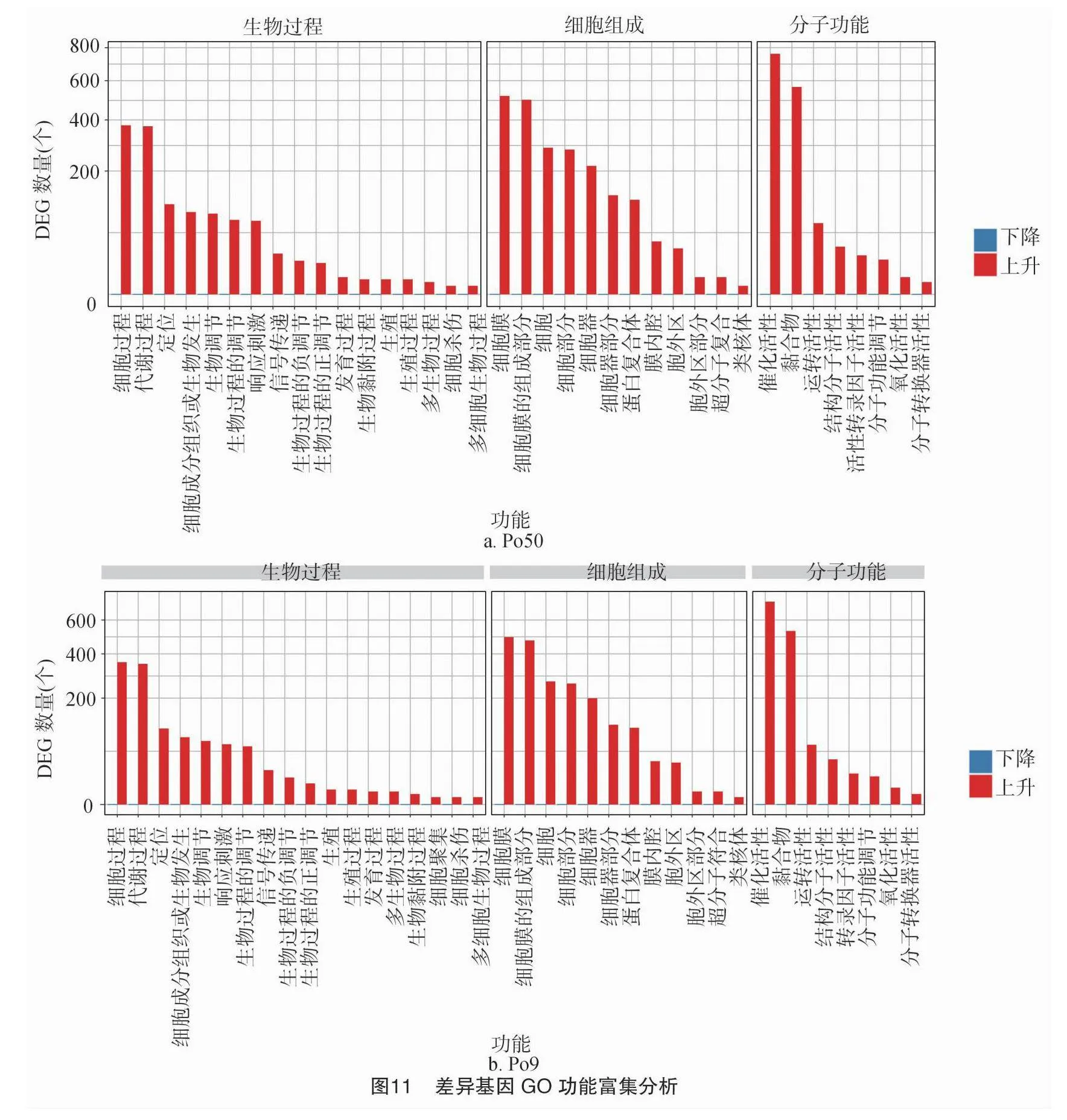
分別篩選出Po50、Po9的差異基因,將差異基因向GO數據庫映射,進行分析,主要富集在生物過程、分子功能和細胞組成相關的3個 GO 類別中,Po50、Po9在生物過程中參與細胞過程上調表達基因分別有372個和349個,代謝過程上調表達基因分別有357個和376個;在細胞組分過程中細胞膜上調表達基因分別有517個和494個,細胞膜組成過程中上調表達基因分別有498個和474個;在分子功能中催化活性上調表達基因分別有761個和725個,黏合物上調表達基因分別有566個和494個(圖11)。
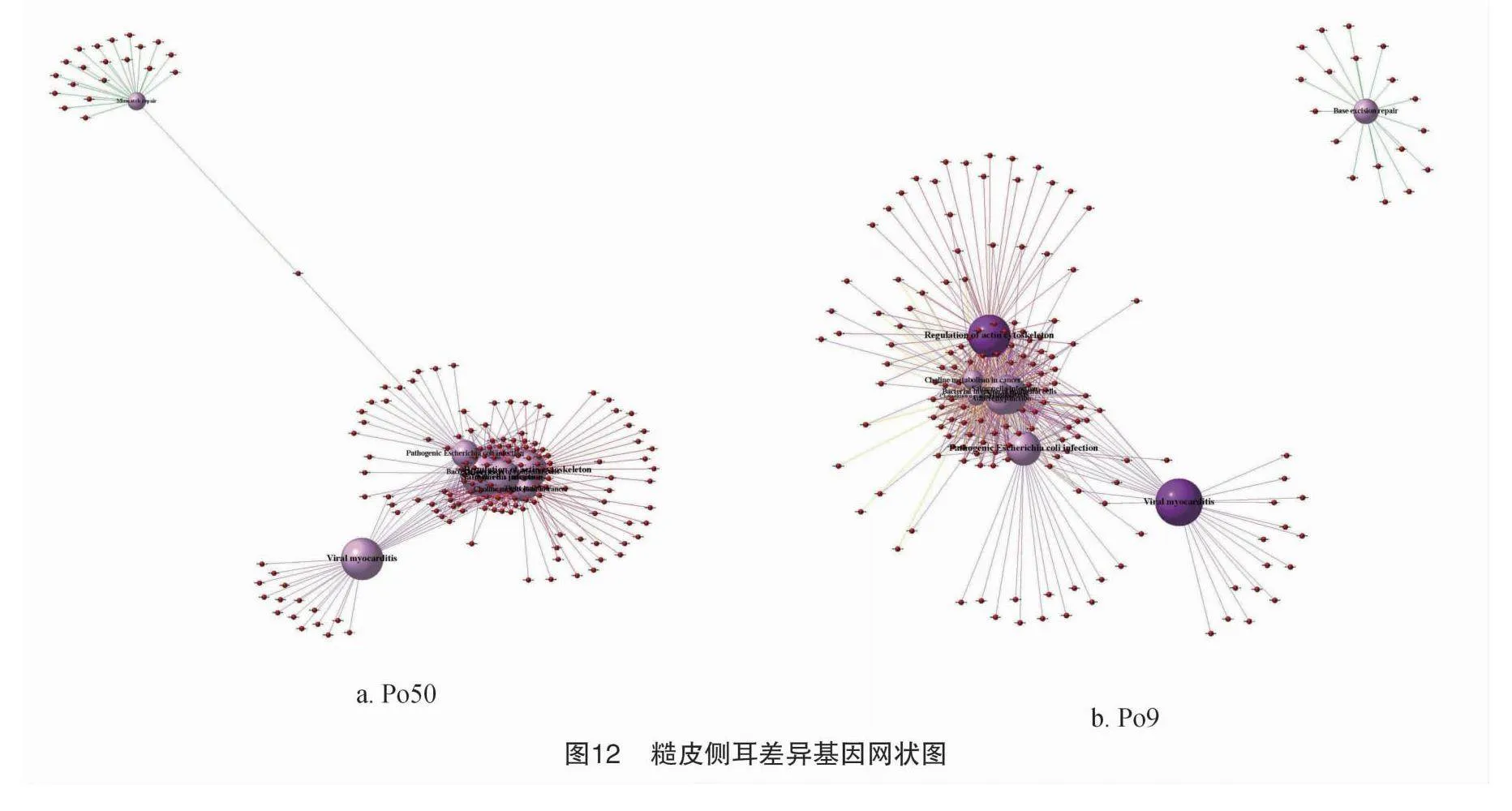
3.1 不同差異基因indel網狀圖
Po50選擇10個顯著的代謝通路如圖12所示:肌動蛋白細胞骨架調控(regulation of actin cytoskeleton)由PC9H_000055等102個基因控制;志賀菌病的致病過程(shigellosis)由PC9H_000055等90個基因控制;致病性大腸桿菌感染(pathogenic escherichia colii nfection)由PC9H_000055等92個差異基因控制;錯配修復(mismatch repair)由PC9H_bMkmRpjPmsYcWVy01BjqIt0/ln7g3cTLk74CKl8Qbm8=000717等21個差異基因控制。附著連接(adherens junction)由PC9H_000055等67個差異基因控制;表皮細胞細菌入侵(bacterial invasion of epithelial cells)由PC9H_000055等71個差異基因控制;癌癥中的膽堿代謝(choline metabolism in cancer)由PC9H_000055等76個差異基因控制,其中還有緊密連接(tight junction)、沙門氏菌感染(salmonella infection)病毒性心肌炎(viral myocarditis)是個顯著通路關系網狀圖。
Po9選擇10個顯著的代謝通路:沙門氏菌感染由PC9H_000055等68個差異基因控制;肌動蛋白細胞骨架調控由PC9H_000055等101個差異基因控制;志賀菌病的致病過程由PC9H_000055等84個基因控制;附著連接由PC9H_000055等65個差異基因控制;病毒性心肌炎由PC9H_000271等30個差異基因控制;癌癥中的膽堿代謝由PC9H_000055等71個差異基因控制;致病性大腸桿菌感染,由PC9H_000249等89個差異基因控制;表皮細胞細菌入侵由PC9H_000249等67個差異基因控制;趨化因子信號通路由PC9H_000249等67個差異基因控制。
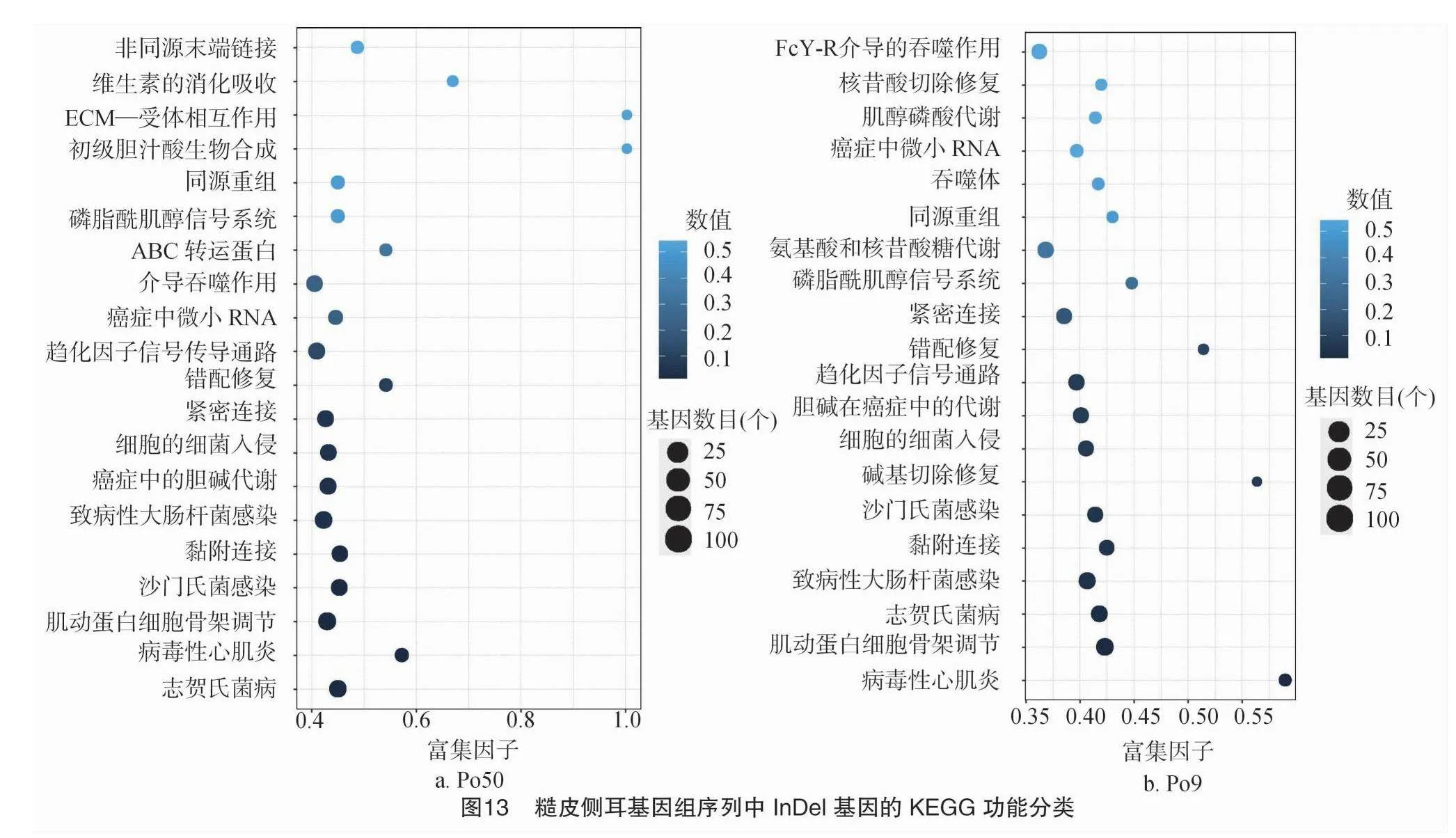
3.2 不同糙皮側耳差異基因的KEGG代謝通路富集分析
將糙皮側耳差異基因通過KEGG數據庫進行Pathway分析,選取KEGG數據庫里的真菌類,選取顯著的通路制作散點圖。對所得到的轉錄本進行驗證和注釋并篩選出的差異基因進行富集通路注釋。將生物代謝通路分為6個類別:細胞過程(cellular processes)、生物體系統(organismal systems)、環境信息處理(environmental information processing)、遺傳信息處理(genetic information processing)、人類疾病(human diseases)、新陳代謝(metabolism)。這些差異基因被分配到7個第1層級通路途徑第2層44個KEGG通路途徑,其中差異基因大部分富集在新陳代謝、疾病、生物體系統以及細胞過程上,其次是生物系統以及環境信息處理上,其中氨基酸和核苷酸糖代謝、緊密連接、白細胞骨架調節和附著連接在新陳代謝、細胞過程占主導地位(圖13)。
4 討論與結論
全基因組重測序已經廣泛應用到人類、動物、植物的基因組和轉錄組分析中[9-12],然而全基因組重測序技術在食用菌中應用較少,通常是應用重測序技術進行ISSR分子標記的開發,在SNP以及InDel標記研究較少[13-15]。
本試驗通過對白色變異菌株進行全基因組重測序從基因組水平精準快速地挖掘了大量的單核苷酸多態性位點SNP,插入缺失位點InDel分析不同個體間的結構差異。Illumina PE150測序平菇白色變異菌株獲得 1 763 Mb高質量數據clean data、平菇黑色出發菌株獲得 1 779 Mb高質量數據 clean data、Po9獲得2 519 Mb高質量數據。在進行全基因組測序時還會檢測到低質量的數據,這些低質量數據會影響基因組的分析,因此基因型對于基因組數據開發SNP以及InDel十分重要,所以重測序數據質量評估至關重要。重測序數據與參考數據樣品對比平菇白色變異菌株Po50檢測到的reads數目為11 754 734個,黑色平菇Po51共檢測到reads數目為11 862 522個,Po9菌株共檢測到16 799 756個 reads。分別檢測到Po51 SNP突變總數1 504 391、1 501 877、1 371 818個。Po50在染色體503 156.1上SNP數目最多。通過突變頻譜分析發現其中T∶A→C∶G、C∶G→A∶T類型的突變較多,SNP變異主要集中于發生在外顯子區域。InDel變異主要集中于發生在基因上游外顯子區域。Po50、Po9測基因進行GO功能注釋,Po50共注釋到5 760個基因,注釋到38個類別,其中CC數量最多,注釋到12個條目,總數為2 433個。將糙皮側耳差異基因通過KEGG數據庫進行Pathway分析,可將生物代謝通路分為6個類別:細胞過程、生物體系統、環境信息處理、遺傳信息處理、人類疾病、新陳代謝。這些差異基因被分配到7個第1層級通路途徑第2層44個KEGG通路途徑[16-18]。本試驗通過重測序技術對平菇白色變異菌株進行鑒定,然而定位到導致顏色變異的基因還有待進一步研究。
參考文獻:
[1]聶興華,張 煜,劉 松,等. 基于基因組重測序的野生板栗遺傳特征和分類地位研究[J]. 園藝學報,2023,50(8):1622-1636.
[2]沈秀芬,章爐軍,張美彥,等. 利用InDel標記分析中國香菇菌株的遺傳多樣性與群體結構[J]. 菌物學報,2021,40(9):2266-2281.
[3]侯炳豪,高 婷,魏月德,等. 基于高深度基因組重測序的‘鐵觀音’茶樹無性繁殖后代遺傳變異研究[J]. 園藝學報,2023,50(7):1505-1517.
[4]Zhao W,Fan L,Wu W J,et al. Re-sequencing and transcriptomic analysis reveal differential expression patterns and sequence variation in glucosyltransferase gene related to anthocyanin biosynthesis in walnut (Juglans regia L.)[J]. Scientia Horticulturae,2023,317:112077.
[5]Zhang Q P,Zhang Y P,Liu W S,et al. Re-sequencing and morphological data revealed the genetics of stone shell and kernel traits in apricot[J]. Frontiers in Plant Science,2023,14:1196754.
[6]Kim J Y,Hwang J E,Eo S H,et al. Development of InDel markers for interspecific hybridization between hill pigeons and feral pigeons based on whole-genome re-sequencing[J]. Scientific Reports,2022,12(1):22618.
[7]Shan T F,Li Y Q,Pang S J. Identification of a genomic region linked with sex determination of Undaria pinnatifida (Alariaceae) through genomic resequencing and genetic linkage analyses of a segregating gametophyte family[J]. Journal of Phycology,2023,59(1):193-203.
[8]Cheng Q,Sun L,Qiao H,et al. Loci underlying leaf agronomic traits identified by re-sequencing celery accessions based on an assembled genome[J]. iScience,2022,25(7):104565.
[9]劉曉雪,王 強,張彬彬,等. 基于酯酶和ISSR技術的平菇單核菌株遺傳多樣性分析[J]. 江蘇農業科學,2022,50(9):27-32.
[10]ShresthaS,Fu Y Q,Michael V N,et al.ftXyIQGdctfetEbRlIEYW7VWtLLRoIhM1OdUDfBZkTI= Whole genome re-sequencing and bulk segregant analysis reveals chromosomal location for Papaya ringspot virus W resistance in squash[J]. Frontiers in Plant Science,2022,13:848631.
[11]Zhao W,Zhang Y P,Zhang J P,et al. QTL mapping by whole genome re-sequencing and analysis of candidate genes for salt tolerance in linseed (Linum usitatissmum L.)[J]. Oil Crop Science,2022,7(2):80-85.
[12]刁興旺,吳莉君,何 紅,等. 芒果炭疽病抗感品種全基因組重測序分析[J]. 江蘇農業科學,2022,50(23):55-61.
[13]Qing J,Meng Y D,He F,et al. Whole genome re-sequencing reveals the genetic diversity and evolutionary patterns of Eucommia ulmoides[J]. Molecular Genetics and Genomics:MGG,2022,297(2):485-494.
[14]Song Z,Zhang Z,Dong J C,et al. Mapping immature fruit colour‐related genes via bulked segregant analysis combined with whole‐genome re‐sequencing in pepper (Capsicum annuum)[J]. Plant Breeding,2022,141(2):277-285
[15]黃平仙,高永明,劉乃新,等. 基于全基因組重測序技術分析甜菜InDel標記[J]. 中國糖料,2020,42(3):1-6.
[16]Wassana K,Pumipat T,Orarat M,等. 基于SNPs全基因組測序技術對泰國辣椒地方品種遺傳多樣性分析和辣椒素含量關聯分析[J]. 辣椒雜志,2019,17(2):37-46.
[17]宋海巖,孫淑霞,李 靖,等. 基于SSR標記檢測與重測序技術的3個李品種鑒定與遺傳背景簡析[J]. 中國南方果樹,2023,52(3):94-101.
[18]李 淦. 基于全基因組重測序解析中國甜柿遺傳多樣性[D]. 武漢:華中農業大學,2022.
