1株西藏來源鏈霉菌10-37的次級代謝產物研究
2024-12-17 00:00:00王嘉涵常珊珊陳銘旭何寧王夢源解云英袁麗杰
中國抗生素雜志 2024年11期
摘要: 目的 對1株西藏來源鏈霉菌10-37的次級代謝產物進行研究。方法 在分子網絡指導下,利用HW40 C凝膠柱、Flash快速液相分離制備色譜儀、半制備型高效液相色譜等方法對化合物進行分離純化,運用核磁共振、高分辨液質聯用技術對單體化合物進行結構鑒定,最后利用微量肉湯稀釋法進行抗菌活性測定。結果 從鏈霉菌10-37的菌絲體提取物中分離得到了4個代謝產物,包括2個新化合物oxazolomycin A3(1)和A4(2),以及2個已知化合物oxazolomycin A2(3)和oxazolomycin B(4)。其中化合物1和2為化合物3的一對幾何異構體,它們三烯部分的構型分別為(4’E,6'E,8'E)(1)、(4’Z,6'E,8'E)(2)、(4’Z,6'Z,8'E)(3)、(4’E,6'E,8'E)(4)。結論 西藏特境的地理優勢結合分子網絡分析的策略,可以更高效地挖掘新結構次級代謝產物。
關鍵詞:鏈霉菌;次級代謝產物;結構鑒定;分子網絡
中圖分類號:R978.1 文獻標志碼:A
Secondary metabolites of Streptomyces sp. 10-37 from Xizang
Abstract Objective This research studied the secondary metabolites of Streptomyces sp. 10-37 collected in Xizang. Methods Under the guidance of the molecular network, the compounds were separated and purified by HW40 C gel columns, flash rapid liquid separation and preparation chromatographs, semi-preparative high-performance liquid chromatography and other methods. The structures of the purified compounds were identified by nuclear magnetic resonance and high-resolution liquid chromatography-mass spectrometry. Finally, the antibacterial activity was determined by the microbroth dilution method. Results Four metabolites were isolated and identified from the mycelium extract of Streptomyces sp. 10-37, including two new compounds, oxazomycin A3 (1) and A4 (2), as well as two known compounds, oxazomycin A2(3) and oxazomycin B(4). Compounds 1 and 2 were a pair of geometric isomers of 3, with the configurations of their diene moieties being (4'E, 6'E, 8'E)(1), (4'Z, 6'E, 8'E)(2), (4'Z, 6'Z, 8'E) (3), and (4'E, 6'E, 8'E)(4), respectively. Conclusion The geographical advantages of Xizang's special environment, combined with the strategy of molecular network analysis, can better excavate new secondary metabolites.
Key words Streptomyces; Secondary metabolites; Structural identification; Molecular network
微生物天然產物因其獨特的結構和豐富的生理活性一直是藥物研發的重要源泉,在抗生素研發領域更是發揮著不可估量的作用[1]。隨著人們對普通環境下微生物的不斷挖掘,發現新骨架天然產物的難度不斷增加,人們開始把注意力投向極端特殊的自然環境,針對“新產地、新菌種、新基因、新產物、新用途”的理念[2]尋找具有潛在藥用價值的新菌種資源。西藏山南地區多樣的地形地貌使其形成了獨特的高原溫帶干旱性氣候,低溫、少雨、高輻射、低氣壓和低氧等獨特的氣候特點[3]孕育了獨特的微生物資源[4]。
由于微生物天然產物的基數不斷擴大,重復發現日益嚴重。為了解決天然產物重復發現的問題,人們基于生物信息學和化學信息學開發了多種方法和工具。目前應用最廣泛的是Dorrestein實驗室開發的全球天然產物社交分子網絡平臺(GNPS)[5-6],其基于二級質譜的相似性將結構相似的化合物聚集成簇,同時通過與多種數據庫[7]中已知化合物的參考二級圖譜進行比對而達到排重的目的[8]。該工具提高了天然產物的早期排重效率,使分離工作可以更有目的性地進行。
基于此,本研究利用分子網絡在采集的西藏特境樣品中優選出有潛力的鏈霉菌10-37,對其進行了深入的研究。通過分子網絡分析快速鎖定目標代謝產物,對其進行分離鑒定,共得到4個化合物,其中化合物1和2為一對新的幾何異構體。
1 材料與方法
1.1 材料
1.1.1 儀器與試劑:
儀器:電子分析天平AB204-E(瑞士Mettler公司);高壓蒸汽滅菌鍋(日本Sanyo公司);無菌超凈工作臺YT-CJ-1D(北京亞泰科隆);恒溫培養箱SHH-C3000 serials(上海合恒儀器設備有限公司);恒溫搖床(上海捷呈實驗儀器有限公司);臺式離心機(美國Sigma公司);小型高速離心機(德國Eppendorf公司);超聲波清洗器(昆山市超聲儀器有限公司KQ5200型);干式氮吹儀(北京天林恒泰科技有限公司);旋轉蒸發儀(EYELA OSB-220低溫冷凍循環裝置、Buchi NS45/40);Flash快速液相分離制備色譜(Combi Flash);冷凍干燥機(CHRIST ALPHA 2-4 LSC);循環水式真空泵SHZ-B-III(鄭州恒巖儀器有限公司);高效液相色譜儀(Agilent Technologies 1290 series);核磁共振波譜儀Bruker AVIIIHD-600MHz(美國Bruker公司);高分辨液質聯用儀器(Waters? UPLC-Xevo? G2-S Qtof)。
分離填料及試劑:HW40 C凝膠(日本Toyopearl公司);Xbridge? Prep C18(5 μm, 250 mm×10 mm)半制備色譜柱;ACQUITY UPLC CSHTM C18柱(1.7 μm,
2.1 mm×100 mm);ODS填料(日本YMC);分析級乙醇(北京通廣精細化工廠);LC-MS級甲酸、甲醇及乙腈(北京百靈威科技有限公司);色譜級甲醇及乙腈(上海星可高純溶劑有限公司);制備級甲酸、甲醇及乙腈(上海星可高純溶劑有限公司);DMSO-d6(Cambridge Isotope Laboratories);實驗用水(娃哈哈公司)。
培養基:ISP-2液體培養基(酵母浸粉4.0 g,麥芽浸粉10.0 g,葡萄糖4.0 g,去離子水1 L,PH 7.4);MHB培養基(牛肉粉2.0 g,酸水解酪蛋白17.5 g,可溶性淀粉1.5 g,去離子水1 L,PH 7.2)。
1.1.2 菌株來源與檢定菌
菌株分離自西藏自治區山南市南部隆子縣高山草甸上的地衣樣品,樣品采集位置為東經91°56'49.58'',北緯28°23'17.70'',海拔4752 m。檢定菌:枯草芽胞桿菌(Bacillus subtilis)CMCC63501、金黃色葡萄球菌(Staphylococcus aureus)ATCC29213、耐甲氧西林金黃色葡萄球菌(methicillin-resistant Staphylococcus aureus,MRSA 臨床株)、大腸埃希菌(Escherchia coli)ATCC25922、產超廣譜β-內酰胺酶的大腸埃希菌(extended-spectrum β-lactamases-producing Escherichia coli, ESBLs, 臨床株)、銅綠假單胞菌(Pseudomonas aeruginosa)ATCC27853。
1.2 方法
1.2.1 菌株發酵
將保存菌株的甘油管從-80 ℃冰箱中取出,通過無菌操作吸取100 μL菌液到ISP-2固體培養基平皿上,涂布棒涂勻后放置在28 ℃溫箱中靜置培養7 d。配制10瓶發酵培養基(100 mL ISP-2液體培養基/500 mL三角燒瓶),每瓶接種約2 cm2大小的菌落,于220 r/min,28 ℃,培養42 h,獲得種子液。配制20瓶發酵培養基(1 L ISP-2液體培養基/5 L三角燒瓶),每瓶接種50 mL種子液,于180 r/min,28 ℃,培養8 d,獲得20 L發酵產物。
1.2.2 分子網絡分析
使用Waters Compression Tool壓縮MS/MS原始.raw文件,后使用UNIFI軟件將壓縮后的.raw文件轉換成.mgf文件,再用MS convert軟件轉化為.mzmL格式文件,利用FileZilla軟件上傳至GNPS-MassIVE數據存儲庫(https://massive.ucsd.edu)。通過GNPS平臺(https://gnps.ucsd.edu)進行MS/MS分子網絡分析,參數為前體離子質量容差0.02 Da;碎片離子質量容差0.02 Da;最小余弦分數值0.65;最小匹配碎片離子數5;最小網格簇數1;其他均為默認參數。最后使用Cytoscape v3.8.0實現結果可視化。
1.2.3 化合物分離純化及結構鑒定
發酵液經4500 r/min離心20 min,菌絲體部分用無水乙醇提取6次,減壓蒸干得到粗提物14.3 g。粗提物用甲醇萃取,離心取上清液過HW40 C凝膠柱(3.2 cm×15 cm),甲醇洗脫,減壓蒸干后得到粗品4.7 g。將其與ODS填料按質量比1:1拌樣,干法上樣,利用Flash進一步分離(條件:RediSep Rf C18 flash column, 130 g;A相:純水;B相:乙腈;0~50 min:10%~55%B;50~65 min:55%~100%B; 65~70 min:100%B;流速25 mL/min)。根據LC-MS檢測結果對相同組分進行合并,減壓濃縮得到21個組分(F1~F21)。
化合物1~3主要存在于F5中,共47.5 mg,利用高效液相色譜儀分離純化(條件:Xbridge? Prep C18半制備柱(5 μm, 250×10 mm);A相:純水(含0.1%甲酸);B相:乙腈(含0.1%甲酸);0~90 min:28%B等度洗脫;流速2.5 mL/min;柱溫30 ℃)得到化合物1~3。化合物4主要存在于F9和F10中,共23.1 mg,利用高效液相色譜儀分離純化(條件:Xbridge? Prep C18半制備柱(5 μm, 250×10 mm); A相:純水(含0.1%甲酸);B相:乙腈(含0.1%甲酸);0~90 min:32%B等度洗脫;流速2.5 mL/min;柱溫30 ℃)得到化合物4。將化合物1~4用純水稀釋5倍之后上樣于ODS柱,純水沖5個柱體積除酸,乙腈沖6個柱體積洗脫,減壓蒸干后得到化合物1(3.6 mg)、化合物2(2.8 mg)、化合物3(10.1 mg)和化合物4(2.1 mg)。
利用高分辨液質聯用儀對化合物進行檢測(條件:ACQUITY UPLC CSHTM C18柱(1.7 μm, 2.1 mm×
100 mm);A相:純水(含0.1%甲酸);B相:乙腈(含0.1%甲酸);0~2 min:10%~32%B;2~9 min, 32%B; 9~11 min, 10%B;流速0.3 mL/min;柱溫30 ℃;質譜采用Fast DDA模式;利用Waters Masslynx (V4.1)軟件進行數據采集),結合1H (600 MHz)和13C NMR(150 MHz)數據對化合物1~4進行結構鑒定。
1.2.4 抗菌活性測定
參照CLSI標準[9],采用微量肉湯稀釋法對化合物3和4進行6種檢定菌的最低抑菌濃度(minimal inhibitory concentration, MIC)測定。使用MHB培養基將0.5 MCF(1×108 CFU/mL)的菌懸液稀釋至5×
105 CFU/mL,96孔板第一孔加入200 μL菌液,2~12孔加入100 μL菌液。待測化合物溶于DMSO配制成12.8 mg/mL母液,第一孔中加入2 μL,混勻,依次進行倍比稀釋,使化合物濃度依次為128、64、32、16、8、4、2、1、0.5、0.25、0.125和0.0625 μg/mL。
以美羅培南和萬古霉素標準品作為陽性對照,DMSO作為陰性對照,MHB培養基為空白對照。
30 ℃恒溫培養12 h后觀察結果,并通過酶標儀讀取各孔的A600吸光值。
2 結果與分析
2.1 GNPS分子網絡分析
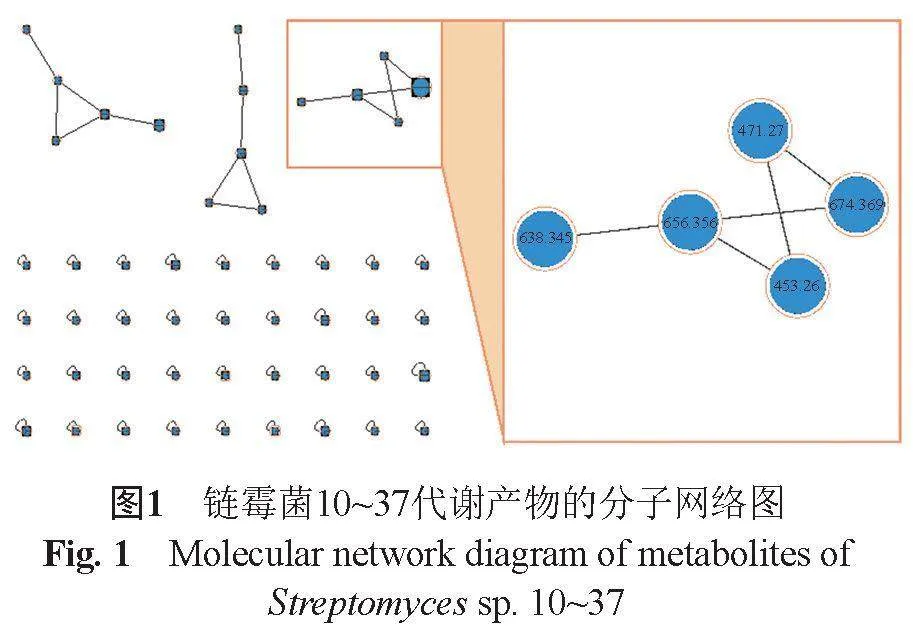
利用GNPS平臺對菌株發酵提取物的LC-MS/MS數據進行分析排重。圖1中,一個節點代表一種化合物(不排除為同分異構體的集合),其上標注的為每個化合物的質荷比。根據分子網絡提供的信息,m/z 674.369的化合物具有較高的強度,在菌株10~37代謝產物中產量占比較大,為該菌株的主要產物;且在GNPS數據庫搜索后發現,該簇并未匹配到與其相似的已知化合物,提示為一類潛在的新化合物,因此,對該簇化合物進行了定向分離。
2.2 結構鑒定
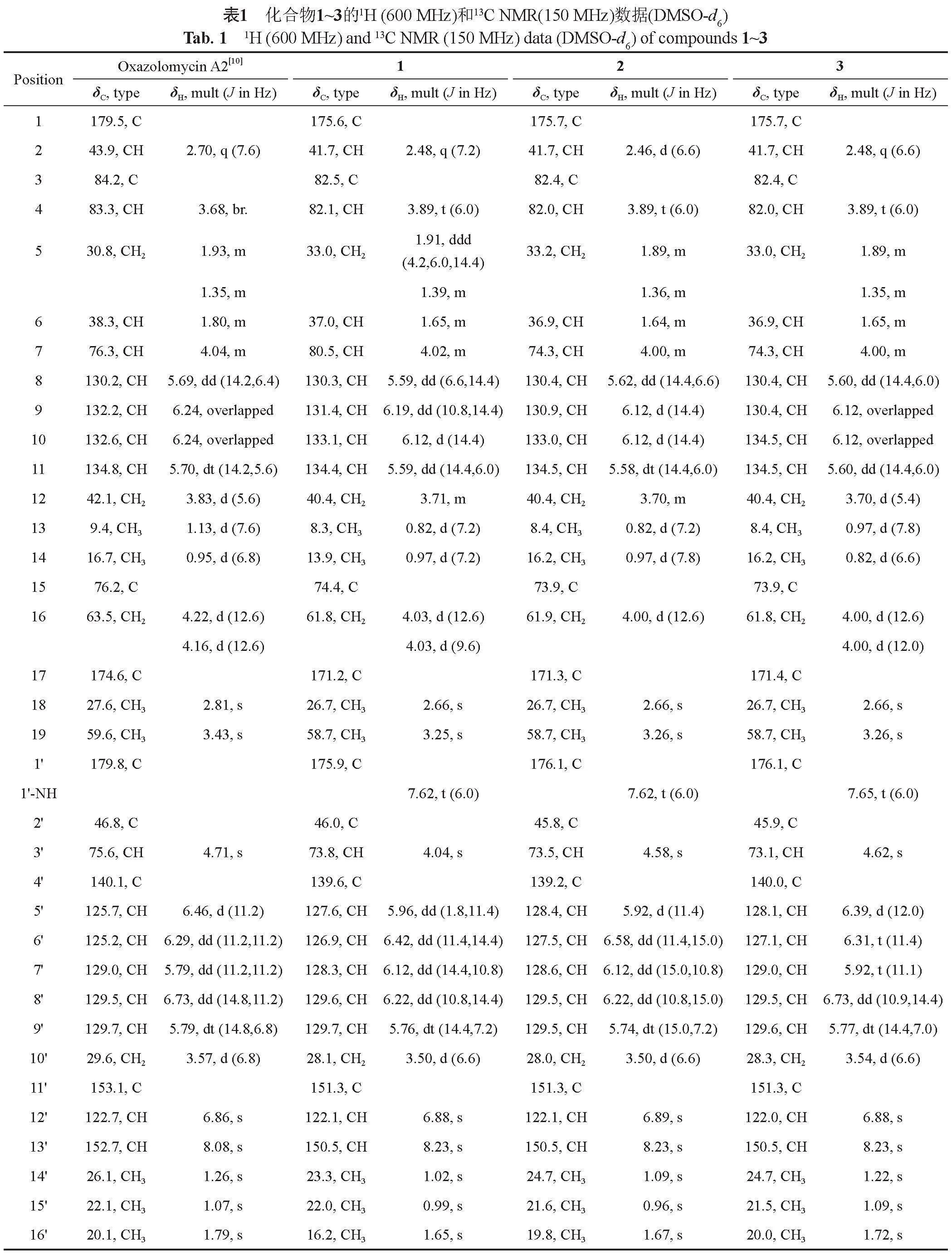
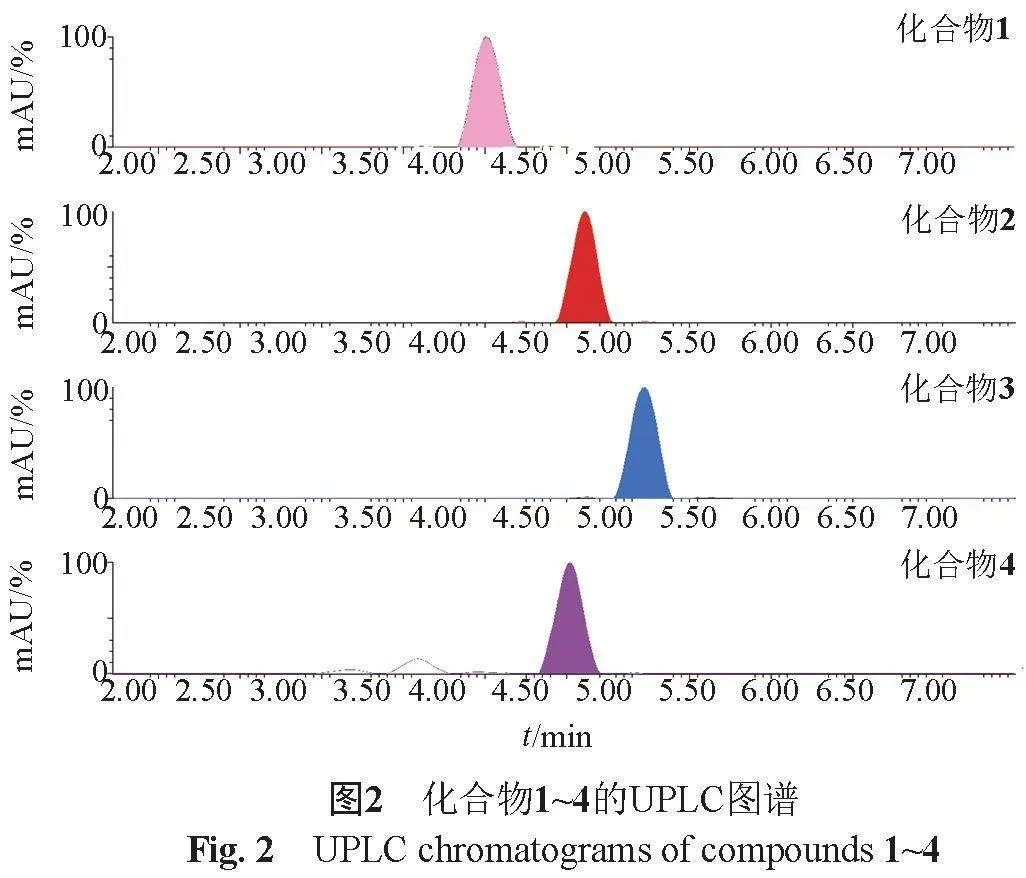
在分子網絡指導下,從鏈霉菌10~37的菌絲體提取物中分離得到了4個化合物,其化合物純度經UPLC檢測如圖2所示。HR-ESI-MS數據表明,化合物1~3具有相同的分子量。UV光譜均在230和275 nm左右顯示出最大吸收值,在265和285 nm處具有兩個肩峰,表明其結構中存在共軛三烯片段。通過微譜檢索,發現化合物1~3與已知化合物Oxazolomycin A2[10]的13C NMR化學位移具有較高相似度,且化合物3的相似性最高(表1),進一步通過1H NMR數據比對確定化合物3即為oxazolomycin A2。
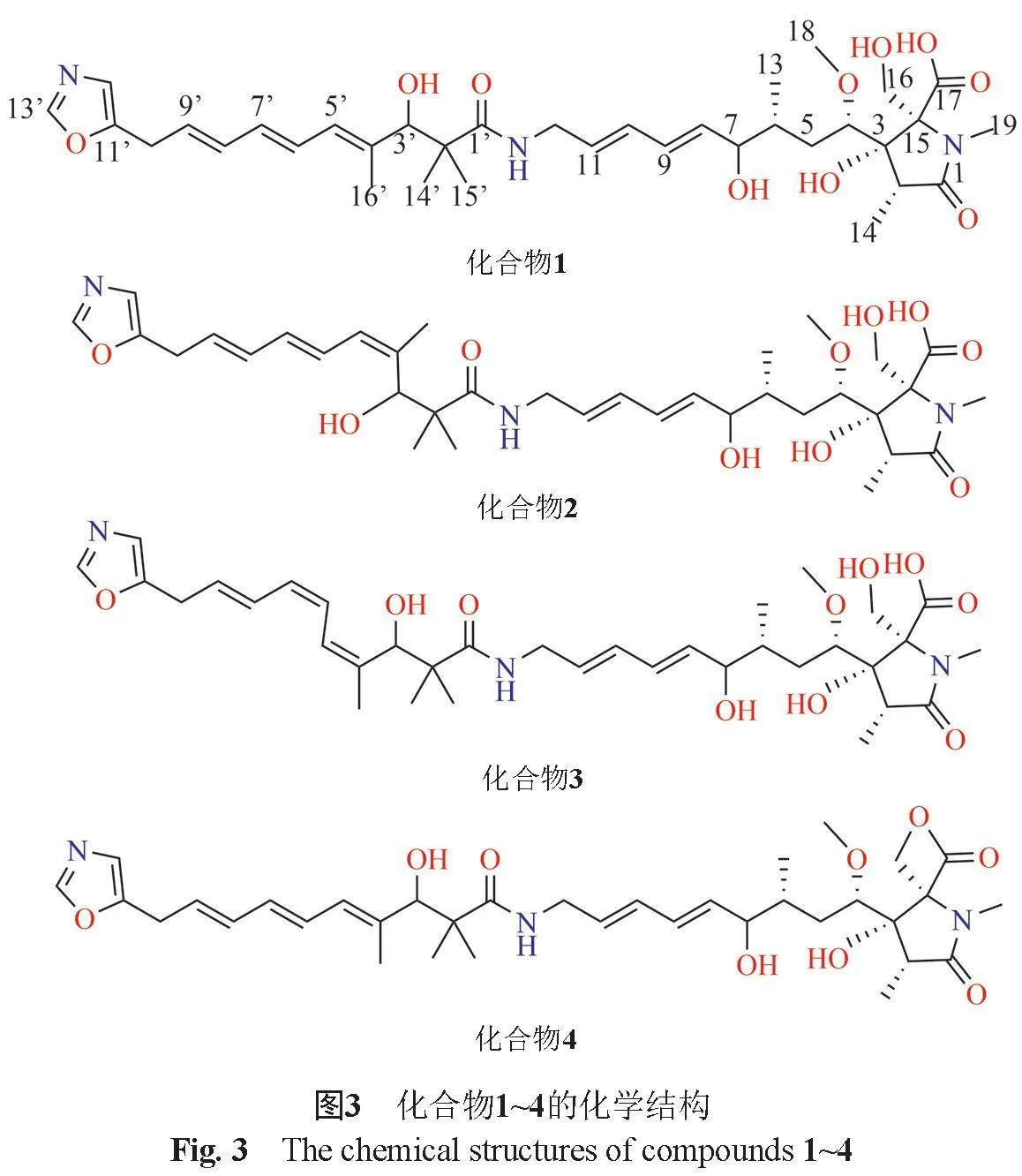
1H NMR的化學位移和耦合常數顯示化合物1~3的差異主要在于三烯部分(H-3', H-4', H-5', H-6', H-7', H-8'和H-9')。通過比較化合物1~3的1H NMR數據,發現化合物1中H-3'的化學位移(δH-3,4.04)與化合物2(δH-3,4.58)和3(δH-3,4.62)的相比向高場位移了約0.6 ppm,由此推斷化合物1中雙鍵Δ4(5)為E構型,而化合物2與3相同為Z構型[10-11]。化合物1和2中H-5’和H-8’化學位移(1:δH-5,5.96,δH-8,6.22;2:δH-5,5.92,δH-8,6.22)與化合物3中的(3:δH-5,6.39,δH-8,6.73)相比向高場位移了約0.5 ppm,因此推測化合物1和2雙鍵Δ6(7)的構型與3中的相反[10],為E構型。H-6'和H-7'之間的耦合常數(1:J6’,7’=14.4 Hz,2:J6’,7’=15.0 Hz,3:J6’,7’=11.1 Hz)進一步證明了以上推測。化合物1~3中H-8'和H-9'之間均具有較大的耦合常數(1:J8’,9’=14.4 Hz,2:J8’,9’=15.0 Hz,3:J8’,9’=14.4 Hz),表明化合物1~3中雙鍵Δ8(9)均為E構型[10]。綜上所述,化合物1~3共軛三烯部分的幾何結構分別為(4’E,6'E,8'E)、(4’Z,6'E,8'E)和(4’Z,6'Z,8'E)(圖3),1和2為一對新的幾何異構體,分別命名為oxazolomycin A3和A4。
化合物1:黃色油狀物;UV λmax(MeOH) 228,278 nm;1H和13C NMR數據(DMSO-d6)參考表1;HR-ESI-MS [M+Na]+ m/z 696.3486(calcd for C35H51N3O10Na, 696.3472, 2.0 ppm)。
化合物2:黃色油狀物;UV λmax (MeOH) 228, 276 nm;1H和13C NMR數據(DMSO-d6)參考表1;HR-ESI-MS [M+Na]+ m/z 696.3486(calcd for C35H51N3O10Na, 696.3472, 2.0 ppm)。
化合物3:黃色油狀物;UV λmax (MeOH) 224, 276 nm;1H和13C NMR數據(DMSO-d6)參考表1;HR-ESI-MS [M+Na]+ m/z 696.3486(calcd for C35H51N3O10Na, 696.3472, 2.0 ppm)。
化合物4:黃色油狀物;UV λmax (MeOH) 223, 276 nm;1H NMR (600 MHz, DMSO-d6) δ 8.23(s, 1H),7.65(t, J=6.0 Hz, 1H),6.89(s, 1H),6.74(dd,J=11.4,14.4 Hz,1H),6.39(d,J=12.0 Hz,1H),6.30(t,J=11.4 Hz,1H),6.20(dd,J=10.2,15.0 Hz,1H),6.11(dd,J=10.2, 15.0 Hz, 1H),5.91(t,J=11.4 Hz,1H),5.77 (dt,J=7.2,6.6 Hz,1H),5.69(ddd,J=6.0,11.4,21.0 Hz,1H),5.49(dd, J=7.8,15.0 Hz,1H),5.42 (d,J=4.8 Hz,1H),4.63(d,J=4.8 Hz,1H),4.16(dd,J=7.2,10.2 Hz, 1H),4.08(d,J=12.6 Hz,1H),3.96(d,J=12.6 Hz, 1H),3.70(t,J=5.4 Hz,2H),3.54(d,J=6.6 Hz,2H),3.51(t,J=5.4 Hz,1H),3.14(s,3H),2.66 (s,3H),2.57(q,J=7.2 Hz,1H),1.85(m,1H),1.72(s,3H),1.68(m,1H),1.22(s,1H),1.09(s,3H),1.02(d,J=7.2 Hz,3H),0.97(s,3H),0.82(d,J=7.2 Hz,3H);13C NMR(150 MHz,DMSO-d6) δ 176.1,175.1,171.2,151.3,150.5,140.0,132.2,131.8,131.8,129.6,129.0,128.1,127.1,124.5,123.5,122.0,84.5,79.8,78.3,74.6,73.0,61.6,55.8,46.0,44.1,40.4,31.8,28.3,27.3,27.1,24.6,21.5,20.0,19.5,9.8:HR-ESI-MS [M+Na]+ m/z 678.3346(calcd for C35H49N3O9Na,678.3366,2.9 ppm)。上述數據與文獻[11]中數據對照基本一致,故確定該化合物4為oxazolomycin B[10]。
2.3 抗菌活性測定
采用微量肉湯稀釋法測定了化合物3和4的最低抑菌濃度,結果顯示,對6種檢定菌均無抑制活性(MIC gt;128 μg/mL)。
3 討論
本研究以分子網絡為導向,從西藏特境鏈霉菌中鑒定了一組oxazolomycin類化合物。Oxazolomycin及其同系物是一個相對罕見的天然產物家族[12-13]。
該類化合物的常見結構特征包括一個獨特的β-內酯-γ-內酰胺螺環結構和一個末端惡唑環,兩部分通過三烯和(E,E)-二烯脂鏈連接[12-13]。到目前為止,該家族成員包括neooxazolomycin[14], oxazolomycins A-F[11, 15],
curromycins A和B(也稱為triedimycins A和B)[16-17],
16-methyloxazolomycin[18-19], KSM-2690 B和C[20], lajollamycins[21-22], bisoxazolomycin A, oxazolomycin A2[10], glaucumycins A和B[15]。此外,還報道了幾種與oxazolomycin相關的化合物,即inthomycins和phthoxazolins,它們只具有惡唑環和三烯片段[23-24]。
Koomsiri等[10]采用紙片法對oxazolomycin A2進行了抗菌活性研究,濃度為30、10、3和1 μg/片,結果顯示對恥垢分枝桿菌、銅綠假單胞菌和枯草芽孢桿菌等均無活性;僅在較高濃度下對大腸桿菌NIHJ(30 μg/片)和金黃色葡萄球菌ATCC6538p(10、30 μg/片)顯示出生長抑制。而本研究中化合物3對測試的大腸埃希菌和金黃色葡萄球菌均沒有顯示出抑制活性(MICgt;128 μg/mL),分析可能是由于與文獻中測試的檢定菌株不同造成的。Kanzakiet等[11]比較了oxazolomycin A-C對根癌土壤桿菌、枯草芽胞桿菌和大腸埃希菌等的生物活性,結果顯示A表現出一定活性,而B和C對測試細菌均無活性(MICgt;100 μg/mL)。考慮到B和C為A的一對幾何異構體,其結構差異同樣在左側三烯片段[11],加之主成分化合物3沒有活性、分離難度大、產量少等原因,因此,沒有對化合物1和2進行抗菌活性測定。
本研究利用分子網絡對同類化合物的聚簇及排重功能,對鏈霉菌10-37代謝產物進行研究,具有較強的靶向性。GNPS中的經典分子網絡方法主要通過與數據庫中已知參考化合物的MS/MS圖譜對比進行排重,當前由于數據庫中參考質譜的數據量有限,限制了其排重能力。這導致我們分離出的主成分(化合物3)仍為已知化合物。任何工具都有其局限性,單一的方法并不能完全解決化合物去重復的問題,如何綜合利用多數據庫和工具高效發現新型天然產物仍是我們需要思考的問題。
參 考 文 獻
Newman D J, Cragg G M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019[J]. J Nat Prod, 2020, 83(3): 770-803.
張兆娟, 楊應昆, 鄒月, 等. 從不同地區網柄菌中分離和純培養的共生放線菌[J]. 菌物研究, 2023, 21(Z1): 131-142.
洛桑旺姆, 赤桑單吉, 拉珍, 等. 西藏山南地區氣候特征分析[J]. 西藏科技, 2010, (8): 55-60.
戚珊珊, 周禮紅, 胡久平, 等. 西藏多地區土壤可培養細菌的分離鑒定及多樣性分析[J]. 西南農業學報, 2017, 30(7): 1629-1635.
Watrous J, Roach P, Alexandrov T, et al. Mass spectral molecular networking of living microbial colonies[J]. Proc Natl Acad Sci USA, 2012, 109(26): E1743-1752.
Quinn R A, Nothias L F, Vining O, et al. Molecular networking as a drug discovery, drug metabolism, and precision medicine strategy[J]. Trends Pharmacol Sci, 2017, 38(2): 143-154.
Wang M, Carver J J, Phelan V V, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking[J]. Nat Biotechnol, 2016, 34(8): 828-837.
Nguyen D D, Wu C H, Moree W J, et al. MS/MS networking guided analysis of molecule and gene cluster families[J]. Proc Natl Acad Sci USA, 2013, 110(28): E2611-2620.
CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard-Ninth Edition[S]. CLSI document M07-A9. Wayne, PA, Clinical and Laboratory Standards Institute. 2012.
Koomsiri W, Inahashi Y, Kimura T, et al. Bisoxazolomycin A: A new natural product from 'Streptomyces subflavus subsp. irumaensis' AM-3603[J]. J Antibiot (Tokyo), 2017, 70(12): 1142-1145.
Kanzaki H, Wada K, Nitoda T, et al. Novel bioactive oxazolomycin isomers produced by Streptomyces albus JA3453[J]. Biosci Biotechnol Biochem, 1998, 62(3): 438-442.
Moloney M G, Trippier P C, Yaqoob M, et al. The oxazolomycins: A structurally novel class of bioactive compounds[J]. Curr Drug Discov Technol, 2004, 1(3): 181-199.
Patrik O, Jozef, G, Eugenie N, et al. The oxazolomycin family: A review of current knowledge[J]. RSC Adv, 2020, 10(67): 40745-40794.
Takahashi K, Kawabata M, Uemura D, et al. Structure of neooxazolomycin, an antitumor antibiotic[J]. Tetrahedron Letters, 1985, 26(8): 1077-1078.
Mu Y, Jiang Y, Qu X, et al. Oxazolomycins produced by Streptomyces glaucus and their cytotoxic activity[J]. RSC Adv, 2021, 11(55): 35011-35019.
Ogura M, Nakayama H, Furihata K, et al. Structure of a new antibiotic curromycin A produced by a genetically modified strain of Streptomyces hygroscopicus, a polyether antibiotic producing organism[J]. J Antibiot (Tokyo), 1985, 38(5): 669-673.
Ikeda Y, Kondo S, Naganawa H, et al. New triene-beta-lactone antibiotics, triedimycins A and B[J]. J Antibiot (Tokyo), 1991, 44(4): 453-455.
Ryu G, Hwang S, Kim S K. 16-Methyloxazolomycin, a new antimicrobial and cytotoxic substance produced by a Streptomyces sp[J]. J Antibiot (Tokyo), 1997, 50(12): 1064-1066.
Ryu G, Kim S K. Absolute stereochemistry determination of 16-methyloxazolomycin produced by a Streptomyces sp[J]. J Antibiot (Tokyo), 1999, 52(2): 193-197.
Otani T, Yoshida K I, Kubota H, et al. Novel triene-beta-lactone antibiotics, oxazolomycin derivative and its isomer, produced by Streptomyces sp. KSM-2690[J]. J Antibiot (Tokyo), 2000, 53(12): 1397-1400.
Manam R R, Teisan S, White D J, et al. Lajollamycin, a nitro-tetraene spiro-beta-lactone-gamma-lactam antibiotic from the marine actinomycete Streptomyces nodosus[J]. J Nat Prod, 2005, 68(2): 240-243.
Ko K, Lee S H, Kim S H, et al. Lajollamycins, nitro group-bearing spiro-β-lactone-γ-lactams obtained from a marine-derived Streptomyces sp[J]. J Nat Prod, 2014, 77(9): 2099-2104.
Tanaka Y, Kanaya I, Takahashi Y, et al. Phthoxazolin A, a specific inhibitor of cellulose biosynthesis from microbial origin. I. Discovery, taxonomy of producing microorganism, fermentation, and biological activity[J]. J Antibiot (Tokyo), 1993, 46(8): 1208-1213.
Shiomi K, Arai N, Shinose M, et al. New antibiotics phthoxazolins B, C and D produced by Streptomyces sp. KO-7888[J]. J Antibiot (Tokyo), 1995, 48(7): 714-719.
