補腦丸質(zhì)量標準研究
2010-07-29 08:21:16楊瑞瑞喬蓉霞杜宏偉李卓敏
中國藥業(yè) 2010年18期
楊瑞瑞,喬蓉霞,杜宏偉,李卓敏
(1.陜西省食品藥品檢驗所,陜西 西安 710061; 2.西安正大制藥有限公司,陜西 西安 710043)
補腦丸是由酸棗仁、當歸、枸杞子、肉蓯蓉、五味子等多味中藥組成的復(fù)方制劑,具有滋補精血、健腦益智、安神鎮(zhèn)驚、化痰熄風的功效,臨床上用于治療迷惑健忘、記憶減退、頭暈、耳鳴、心煩失眠、心悸不寧、癲癇頭痛、神煩胸悶等癥[1]。原質(zhì)量標準中只有當歸、五味子等幾味藥材的顯微特征鑒別。為了更好地控制產(chǎn)品的內(nèi)在質(zhì)量,筆者分別采用薄層色譜(TLC)法和反相高效液相色譜(RPHPLC)法進行了定性定量研究,報道如下。
1 儀器與試藥
Waters-Alliance 2695-2487型(YY01520041103)高效液相色譜儀,蒸發(fā)光散射檢測器 (Alltech ELSD 2000)。酸棗仁皂苷A對照品(批號為 110734-200509)、酸棗仁皂苷 B對照品(批號為1110814-200506)、枸杞子對照藥材(批號為121072-0301)、松果菊苷對照品(批號為111670-200401)、當歸對照藥材(批號為120927-200310),均由中國藥品生物制品檢定所提供;補腦丸樣品及陰性樣品均由西安正大制藥有限公司提供。乙腈為色譜純,水為高純水,其他試劑均為分析純。
2 方法與結(jié)果
2.1 定性鑒別(TLC法)
當歸:取當歸對照藥材1 g,加乙醚20 mL,超聲處理10 min,濾過,濾液揮干,殘渣加乙醇2 mL使溶解,作為對照藥材溶液;取本品適量,除去包衣,研細,取1 g,加乙醚20 mL,超聲處理10 min,濾過,濾液揮干,殘渣加乙醇2 mL使溶解,作為供試品溶液;取不含當歸的陰性樣品1 g,按供試品溶液制備方法制備陰性對照品溶液。吸取上述3種溶液各5 μL,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以正己烷-乙酸乙酯(4∶1)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品溶液色譜中,在與對照藥材溶液色譜相應(yīng)位置上顯相同顏色的熒光斑點,陰性樣品無干擾(圖1 A)。
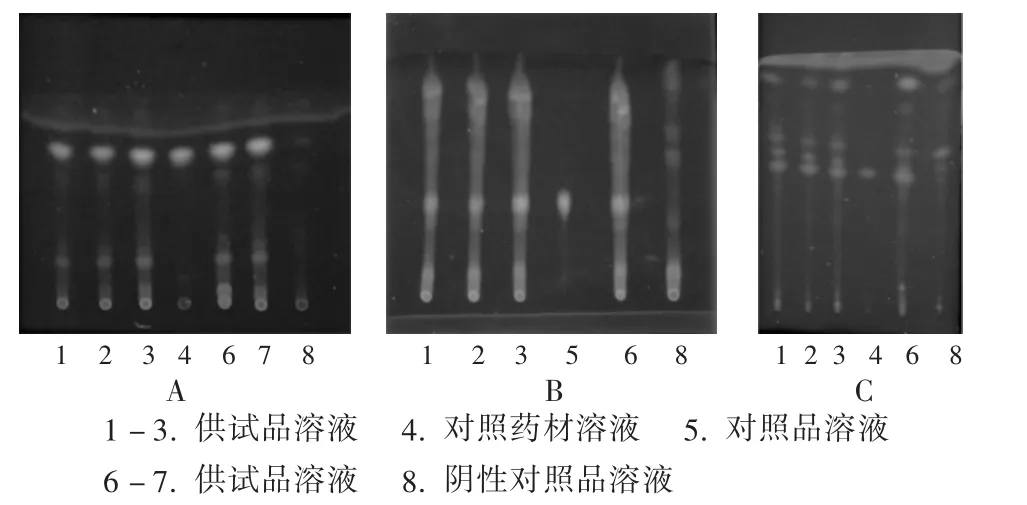
圖1 補腦丸中當歸、肉蓯蓉及枸杞子的薄層色譜圖
肉蓯蓉:取松果菊苷對照品,加甲醇制成每1 mL含0.5 mg的溶液,作為對照品溶液;取本品適量,除去包衣,研細,取1 g,加甲醇20 mL,超聲處理15 min,濾過,濾液濃縮至近干,殘渣加甲醇2 mL使溶解,作為供試品溶液;取不含肉蓯蓉的陰性樣品1 g,按供試品溶液制備方法制備陰性對照品溶液。吸取上述3種溶液各1 μL,分別點于同一聚酰胺薄膜上,以甲醇-醋酸-水(2∶1∶7)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品溶液色譜中,在與對照品溶液色譜相應(yīng)位置上顯相同顏色的熒光斑點,陰性樣品無干擾(圖1 B。)
枸杞子:取枸杞子對照藥材0.5 g,加水50 mL,加熱煮沸15 min,放冷,濾過,濾液用乙酸乙酯25 mL振搖提取,提取液濃縮至約1 mL,作為對照藥材溶液;取本品適量,除去包衣,研細,取5 g,加水50 mL,加熱煮沸15 min,放冷,濾過,濾液用乙酸乙酯25 mL振搖提取,提取液濃縮至約1 mL,作為供試品溶液。取不含枸杞子的陰性樣品5 g,按供試品溶液制備方法制備陰性對照品溶液。吸取上述3種溶液各5 μL,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以甲苯-乙酸乙酯-甲酸(10∶5∶2)為展開劑,展開,取出,晾干,置紫外光燈(365 nm)下檢視。供試品溶液色譜中,在與對照藥材溶液色譜相應(yīng)位置上顯相同顏色的熒光斑點,陰性樣品無干擾(圖1 C)。
酸棗仁:取酸棗仁皂苷A和酸棗仁皂苷B對照品適量,加甲醇分別制成每1 mL含0.5 mg的對照品溶液;取2.2.2項下的供試品溶液,作為供試品溶液,取不含酸棗仁的陰性樣品5 g,按供試品溶液制備方法制備溶液,作為陰性對照品溶液。吸取酸棗仁皂苷A對照品溶液與酸棗仁皂苷B對照品溶液各5 μL,供試品溶液、陰性對照品溶液各10 μL,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以正丁醇-冰醋酸-水(4∶1∶5)的上層溶液為展開劑,展開,取出,晾干,噴以2%香草醛硫酸溶液,在100℃加熱至斑點顯色清晰。供試品溶液色譜中,在與對照品溶液色譜相應(yīng)位置上顯相同顏色的斑點,陰性樣品無干擾(圖2)。
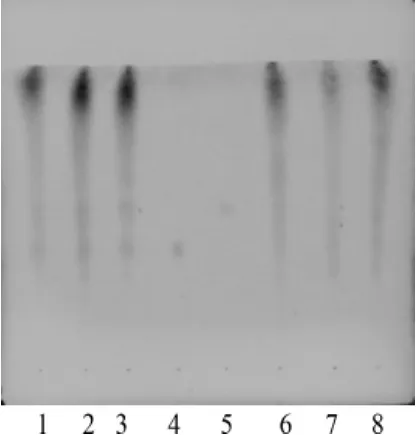
圖2 補腦丸中酸棗仁薄層色譜圖
2.2 酸棗仁皂苷A含量測定(RP-HPLC法)
2.2.1 色譜條件
色譜柱:Kromasil C18柱(250 mm×4.6 mm,5μm);柱溫:30 ℃;流動相:乙腈 -水 (33 ∶67);流速:1.0 mL/min;氣流:2.5 mL/min;漂移管溫度:110℃。
2.2.2 溶液制備
精密稱取酸棗仁皂苷A對照品適量,加甲醇制成每1 mL含0.2 mg的對照品溶液。取本品適量,除去包衣,研細,精密稱取5 g,置索氏提取器中,加乙醚適量回流提取3 h,棄去醚液;藥渣揮盡殘醚,再加甲醇適量回流提取5 h;甲醇提取液置水浴上蒸干,殘渣加水20 mL,分數(shù)次定量轉(zhuǎn)移到分液漏斗中;用水飽和的正丁醇提取5次,每次15 mL;合并正丁醇提取液,用正丁醇飽和的氨水(取40 mL濃氨溶液加水稀釋到100 mL,再用正丁醇飽和,分取下層液)洗滌2次,每次50 mL,棄去洗液,再用正丁醇飽和的水洗滌2次,每次50 mL,正丁醇提取液置水浴上蒸干;殘渣加甲醇溶解,定量轉(zhuǎn)移至5 mL容量瓶中,加甲醇稀釋至刻度,搖勻,作為供試品溶液。取不含酸棗仁的陰性樣品,按供試品溶液制備方法制成陰性對照品溶液。
2.2.3 方法學(xué)考察
專屬性試驗:取2.2.2項下3種溶液,按擬訂的色譜條件進樣測定,結(jié)果見圖3。可見,供試品溶液色譜在與酸棗仁皂苷A對照品相應(yīng)保留時間有一致的色譜峰,陰性對照品溶液無干擾。
線性關(guān)系考察:精密稱取酸棗仁皂苷A對照品 12.45 mg,置 25 mL 量瓶中,加甲醇溶解并稀釋至刻度,搖勻,即得質(zhì)量濃度為 0.498 mg/mL 的對照品溶液,按擬訂的色譜條件分別進樣 1,2,5,10,15,20,25,30,40,50 μL,記錄色譜圖,計算峰面積,以峰面積的對數(shù)值與進樣量的對數(shù)值進行回歸,得回歸方程 Y=1.4102 X+5.7014,r=0.999(n=10)。結(jié)果表明酸棗仁皂苷A進樣量線性范圍是0.498 ~ 24.9 μg。
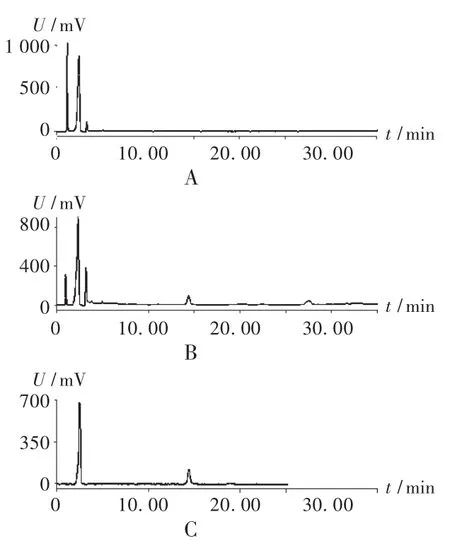
圖3 高效液相色譜圖
精密度試驗:取對照品溶液,重復(fù)進樣6次,依法測定。結(jié)果峰面積的 RSD為4.01%(n=6),表明儀器精密度良好。
穩(wěn)定性試驗:取同一供試品溶液,于24 h內(nèi)分別進樣5次。結(jié)果峰面積的 RSD為4.80%(n=5),表明供試品溶液在24 h內(nèi)基本穩(wěn)定。
重復(fù)性試驗:精密取同一批樣品6份,依法制備供試品溶液并測定。結(jié)果的 RSD為5.33%(n=6)。
加樣回收試驗:精密稱取重復(fù)性試驗樣品細粉共7份,每份2.5 g,分別精密加入酸棗仁皂苷A對照品溶液(0.4265 mg/mL)1 mL,揮干,按供試品溶液制備方法制備溶液,依法測定。結(jié)果見表1。
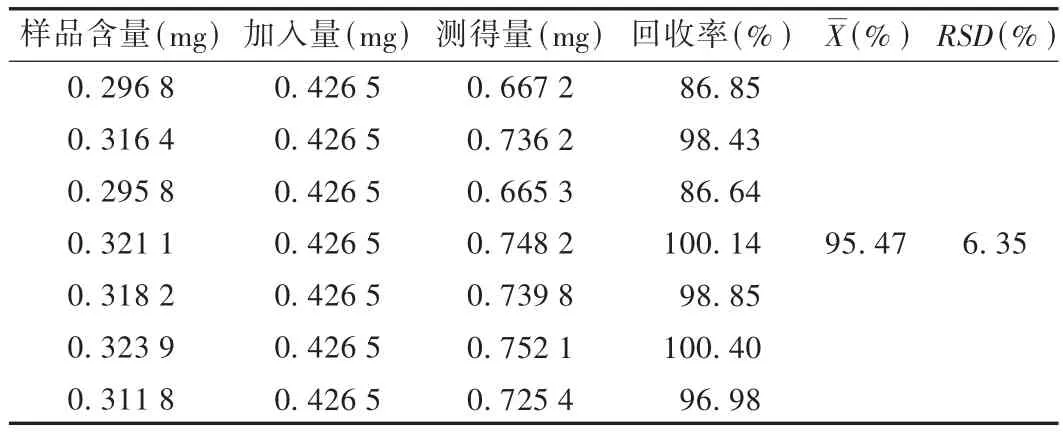
表1 酸棗仁皂苷A加樣回收試驗結(jié)果(n=7)
2.2.4 樣品含量測定
取從市場上收集到的13批樣品,依法制備供試品溶液并測定酸棗仁皂苷A含量。結(jié)果見表2,可見,平均含量為0.1022 mg/g。
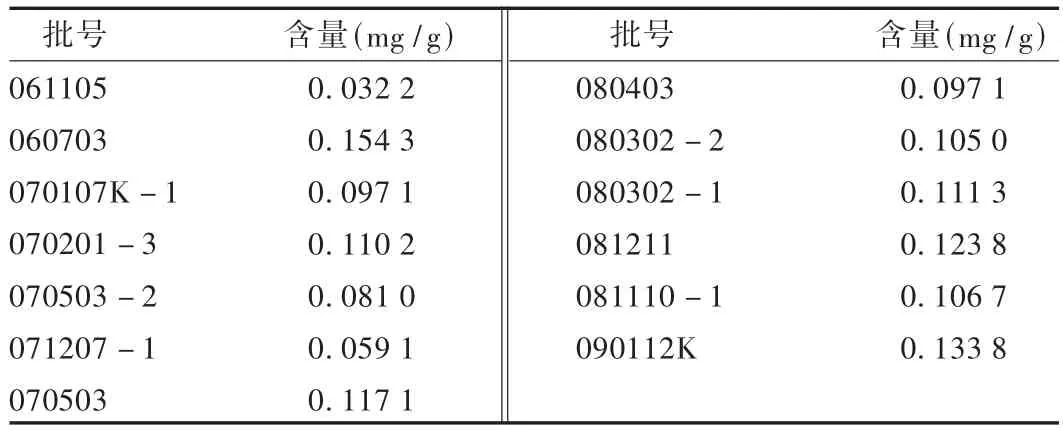
表2 樣品含量測定結(jié)果
3 討論
補腦丸中的酸棗仁[2]具有養(yǎng)心安神的功效,其中所含的酸棗仁皂苷A及酸棗仁皂苷B為有效成分之一,兩成分的含量測定方法多采用薄層掃描法[3],近年來有人采用HPLC法測定。本試驗參考文獻[4-5],用HPLC法對補腦丸主要藥味酸棗仁中的酸棗仁皂苷A進行了含量測定,結(jié)果表明,該方法簡便、準確、重現(xiàn)性好,可有效控制產(chǎn)品質(zhì)量。
在預(yù)試驗中,對供試品溶液制備中的取樣量、提取溶劑、提取時間、除雜溶劑及用量等進行了多因素考察,結(jié)果以乙醚脫脂、甲醇提取、正丁醇萃取、氨溶液除雜質(zhì)為較優(yōu)條件;對色譜條件也進行了優(yōu)化,比較了甲醇-水系統(tǒng)和乙腈-水系統(tǒng),結(jié)果以乙腈-水(33 ∶67)為優(yōu)。
TLC鑒別中,當歸、枸杞子用對照藥材作對照,均能得到滿意的層析結(jié)果;肉蓯蓉因有兩種,選用松果菊苷和毛蕊花糖苷作對照,薄層色譜不完全一致,樣品中在與毛蕊花糖苷斑點相同位置有干擾,故只選松果菊苷作對照;酸棗仁皂苷B在含量測定中量較小,未作為控制指標,故增加在TLC鑒別中。
[1]WS3-B-1949-95,中華人民共和國衛(wèi)生部部頒標準·中藥成方制劑(第十冊)·補腦丸[S].
[2]國家藥典委員會.中華人民共和國藥典(一部)[M].北京:化學(xué)工業(yè)出版社,2005:254.
[3]尹清茹,張永玲.薄層掃描法測定咽炎丸中酸棗仁皂苷的含量[J].中國藥業(yè),1999,8(8):34 -35.
[4]鐘東會,雷根虎,郭五保,等.RP-HPLC測定酸棗仁中的酸棗仁皂苷A的含量[J].中成藥,2004,26(11):963-964.
[5]李會軍,李 萍.高效液相色譜法蒸發(fā)光散射檢測器測定酸棗仁皂苷A及B的含量[J].藥物分析雜志,2000,20(2):82-84.