HPLC法測定黃藤素微丸膠囊中鹽酸巴馬汀的含量*
2012-01-15 06:42:24王京昆
云南中醫中藥雜志 2012年4期
劉 波, 楊 惠, 王京昆
(云南省藥物研究所, 云南 昆明 650111)
黃藤素微丸膠囊是由防已科植物黃藤(Fibraurea recisa Pierre.)的莖中提取的生物堿黃藤素(也稱鹽酸巴馬汀、鹽酸棕櫚堿或掌葉防已堿)加工而成的微丸膠囊劑。有清熱解毒的作用,臨床用于婦科炎癥、菌痢、腸炎、呼吸道及泌尿道感染、外科感染等。
微丸屬于多單元給藥系統,能將一個劑量的藥物分散在千百個微小圓形隔室內,用藥后藥物質點廣泛分布在胃腸道粘膜表面,使藥物吸收完全,從而提高生物利用度。由于微丸粒徑小,受消化道輸送食物節律影響小,所以藥物在體內很少受到胃排空功能變化的影響,在體內的吸收具有良好的重現性。同時,微丸的粒徑均勻,流動性好,不易壓碎,易充填,是一種理想的劑型。
本文采用高效液相色譜法測定黃藤素微丸膠囊中的鹽酸巴馬汀,結果表明該方法簡便、準確、重復性好,可作為黃藤素微丸膠囊含量測定方法。
1 儀器與試藥
實驗用儀器為美國Agilent公司1100型HPLC儀。包含四元泵;紫外檢測器;Agilent Technologies ChemStation色譜工作站;鹽酸巴馬汀對照品:由中國藥品生物制品檢定所提供,批號0732-200005(供鑒別用,經面積歸一化法計算,含量為98.91%)。黃藤素微丸膠囊由云南省藥物研究所藥物制劑研究室提供。乙腈為色譜純,其余試劑均為分析純。
2 方法與結果
2.1 色譜條件 色譜柱:ZorBax Eclipse XDB-C18柱(150 mm×4.6 mm,5 μm),乙腈-0.4%磷酸(25:75)為流動相,檢測波長為345 nm,流速為1.0 mL·min-1,柱溫40 ℃。理論塔板數按鹽酸巴馬汀色譜峰計算應不低于3000。
2.2 溶液的制備
2.2.1 對照品溶液的制備 精密稱取鹽酸巴馬汀對照品適量,加1%鹽酸甲醇液制成每1 mL含0.1 mg的溶液,用0.45 μm微孔濾膜濾過,即得。
2.2.2 供試品溶液的制備 取本品內容物,研細,精密稱取約50 mg,置25 mL容量瓶中,加1%鹽酸甲醇液20 mL,超聲處理20 min,冷卻至室溫,加1%鹽酸甲醇液至刻度,濾過,取續濾液1 mL,置25 mL容量瓶中,加1%鹽酸甲醇液至刻度,用0.45 μm微孔濾膜濾過,即得。
2.2.3 陰性樣品溶液的制備 取陰性樣品內容物,研細,精密稱取約50 mg,置25 mL容量瓶中,加1%鹽酸甲醇液20 mL,超聲處理20 min,冷卻至室溫,加1%鹽酸甲醇液至刻度,濾過,取續濾液1 mL,置25 mL容量瓶中,加1%鹽酸甲醇液至刻度,用0.45 μm微孔濾膜濾過,即得。
2.3 系統適應性實驗 在2.1色譜條件下,分別取對照品溶液、供試品溶液,以及不含鹽酸巴馬汀的陰性樣品溶液10 μL進樣分析,結果見圖1,供試品色譜中,在與對照品色譜圖相應的保留時間上,有相同的色譜峰,而陰性對照在此無干擾。
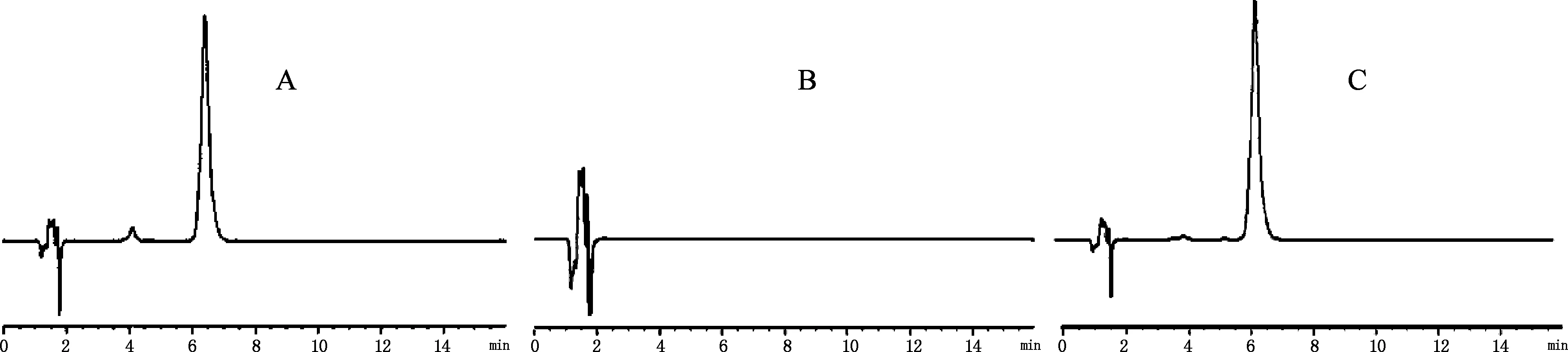
圖1HPLC色譜圖
A.樣品 B.空白樣品 C.鹽酸巴馬汀對照品
3 方法學考察
3.1 線性關系考察 精密稱取鹽酸巴馬汀對照品6.55 mg置25 mL容量瓶中,加甲醇稀釋至刻度,搖勻,精密吸取對照品溶液0.5、1、2、3、4、5、6 mL分別置10 mL容量瓶中,用1%鹽酸甲醇液定容至刻度,搖勻。精密吸取10 μL,分別進樣,檢測,以進樣量(μg)為橫坐標X,峰面積為縱坐標Y,繪制標準曲線,得回歸方程:Y=376 13.651X-18.300,r=0.999 84,結果鹽酸巴馬汀在0.131~1.572 μg范圍內呈良好的線性關系。
3.2 精密度試驗 精密吸取上述對照品溶液10 μL,重復進樣6次,以峰面積計算,結果鹽酸巴馬汀的RSD為0.13%(n=6),表明本法的精密度較好。
3.3 重現性實驗 取同一批號樣品6份,按供試品溶液的制備方法制備,進樣10μl測定,以峰面積計算鹽酸巴馬汀的含量為23.34%,測定結果的RSD為1.06%(n=6),表明本法的重現性較好。
3.4 穩定性試驗 取同一批供試品,分別于0、1、2、3、6、12、24 h進樣,進樣體積10 μL,以峰面積計算,結果RSD為0.28%(n=7),所得的樣品中鹽酸巴馬汀峰面積基本一致,說明樣品在24 h內基本穩定。
3.5 加樣回收率試驗 準確稱取已知含量的黃藤素微丸膠囊樣品,精密加入適量的鹽酸巴馬汀對照品溶液,照色譜條件測定,計算回收率,結果見表1,RSD=2.46%(n=9)。
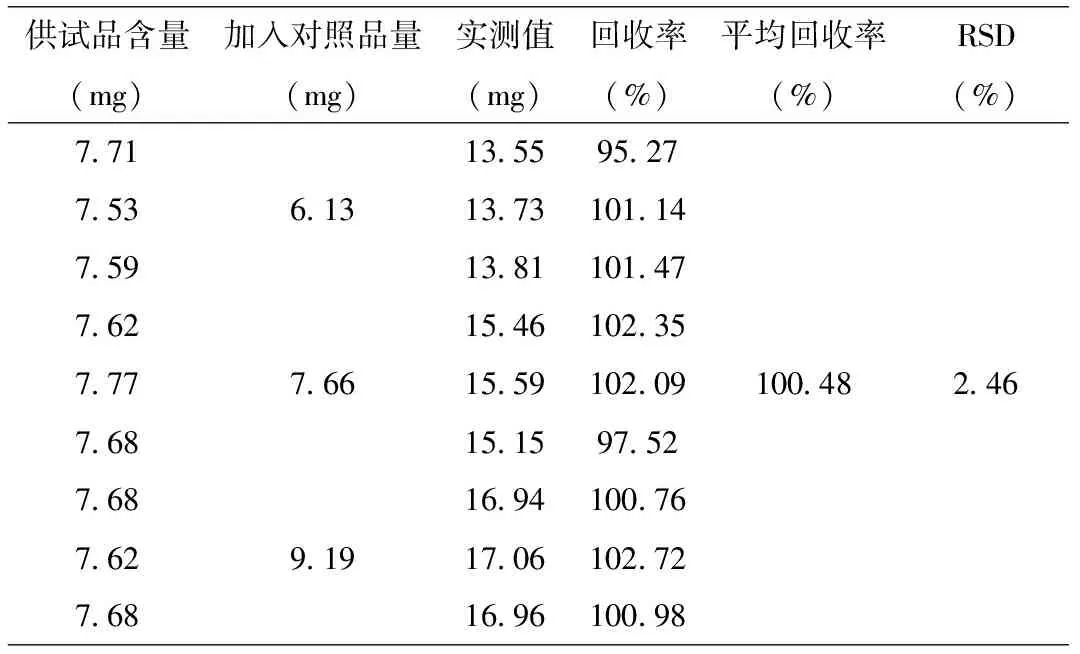
表1 加樣回收率試驗結果
3.6 樣品的含量測定 按鹽酸巴馬汀含量測定方法測定3批樣品,結果三批樣品的含量為平均每粒含鹽酸巴馬汀100.71、100.37、100.43 mg,根據以上結果,將黃藤素微丸膠囊中鹽酸巴馬汀的含量參照膠囊制劑的裝量差異定為:每粒含鹽酸巴馬汀為90~110 mg。
4 討論
4.1 前處理方法的選擇 選用同一批黃藤素微丸膠囊,取內容物研細后,精密稱定,平行稱取5份,溶劑選用甲醇、1%鹽酸甲醇液,分別選用下列五種常用提取方法提取:(1)(2)(3)置25 mL容量瓶中,加1%鹽酸甲醇液20 mL,分別超聲處理10、20、30 min,放冷至室溫,加1%鹽酸甲醇液定容至刻度,濾過,取續濾液1 mL,置25 mL容量瓶中,加1%鹽酸甲醇液定容至刻度,即得。(4)置25 mL容量瓶中,加甲醇20 mL,超聲處理20 min,放冷至室溫,加甲醇定容至刻度,濾過,取續濾液1 mL,置25 mL容量瓶中,加甲醇定容至刻度,即得。(5)置25 mL容量瓶中,加1%鹽酸甲醇液20 mL,水浴20 min,振搖2~3次,放冷至室溫,加1%鹽酸甲醇液定容至刻度,濾過,取續濾液1 mL,置25 mL容量瓶中,加1%鹽酸甲醇液定容至刻度,即得。
提取后用同一方法進行測定,結果表明,方法(2)、(3)提取率最高且相近,為節約檢測時間故采用方法(2)來處理樣品。
4.2 檢測波長的選擇 通過對鹽酸巴馬汀的甲醇溶液進行全波長掃描,結果鹽酸巴馬汀在265 nm、345 nm、348 nm處有最大吸收,但在345 nm處的吸收值最大,與文獻報導一致1~3,因此,選用345 nm作為檢測波長。
4.3 流動相的選擇 根據查閱的相關文獻資料1~3和質量標準,筆者做了如下的研究:(1)乙腈-水-十二烷基磺酸鈉(30∶70∶0.1 g);(2)乙腈-0.033 mol/L磷酸二氫鉀水溶液(30∶70);(3)乙腈-0.4%磷酸(25∶75)
實驗結果表明,方法(1)流動相主峰有拖尾,色譜峰的對稱性較差,方法(2)、方法(3)色譜峰峰型都較好,分離度高,但方法(3)色譜峰的對稱性較好,故將流動相定為乙腈-0.4%磷酸(25:75),能得到較為滿意的色譜峰,提高了測定結果的準確性。
4.4 色譜分離耐用性試驗 考察了DiamonsilTM-C18、ZorBax Eclipse XDB-C18、ZorBar SB-C18三種不同型號的十八烷基硅烷鍵合硅填充柱,都能滿足鹽酸巴馬汀含量測定的條件。
為了得到更好的分離結果,將黃藤素微丸膠囊中鹽酸巴馬汀含量測定的色譜條件定為:以十八烷基硅烷鍵合硅膠為填充劑;乙腈-0.4%磷酸(25:75)為流動相;檢測波長為345nm;理論塔板數按鹽酸巴馬汀色譜峰計算應不低于3000。
[1]金佩芬,繆華蓉,錢亞萍,等.HPLC測定戊己丸中鹽酸小檗堿和鹽酸巴馬汀的含量[J].中成藥,2001.23(1):21.
[2]黃捷,桑彤,覃紅萍.反相高效液相色譜法測定金雞膠囊中鹽酸巴馬汀及鹽酸小檗堿的含量[J].中國醫院藥學雜志,2002.22(12):35.
[3]毛頤晴,彭瑜.高效液相色譜法測定黃藤素片中鹽酸巴馬汀的含量[J].中成藥,2004.7(12):65.
