索拉菲尼對肝癌細胞的抑制作用及其對CDK5表達水平的影響
2012-01-29 02:43:44趙浩亮
中國醫藥科學 2012年17期
關鍵詞:肝癌
劉 沖 趙浩亮
1.太原市中心醫院普外一科,山西太原 030009;2.山西醫科大學第一醫院普外科,山西太原 030001
原發性肝癌(PHC)是臨床上常見的惡性腫瘤之一,其惡性程度高、容易轉移和復發,嚴重威脅著人類的健康,因此尋找一種合理有效的治療方案則具有重大的意義。隨著基因工程技術和現代分子生物學的發展,以免疫治療為基礎的生物治療及分子靶向藥物治療越來越受到關注,并逐漸成為肝癌治療的重要手段,為肝癌的治療提供了新思路[1]。索拉菲尼(sorafenib)是首個口服的多靶點、多激酶抑制劑,作為第1個在肝癌治療中獲得優勢生存的靶向藥物,已被美國食品與藥品管理局(food and drug administration,FDA)確定用于肝癌患者的治療。周期素依賴性激酶5(CDK5)是細胞周期調控網絡的核心蛋白(CDK)家族中的一員,它通過影響細胞增殖和凋亡異常而導致腫瘤的發生發展。本實驗觀察索拉菲尼在體外對人肝癌HepG2細胞株的抑制作用及其對CDK5表達的影響。
1 材料與方法
1.1 材料
1.1.1 接種細胞 人肝癌HepG2細胞株:購自中國科學院上海生命科學研究院細胞庫。
1.1.2 主要試劑 RPMI1640培養基(武漢博士德公司,批號:PYG0035);胎牛血清(杭州四季青公司,批號:040622);二甲基亞楓(DMSO,南京賽泓瑞生物科技有限公司,批號:D-5879)及四甲基偶氮唑鹽(MTT,美國Sigma公司,批號:298-93-1);索拉菲尼(Sorafenib,德國拜耳公司,批號: FS10807 );兔抗人CDK5多克隆抗體和羊抗兔抗體( 二抗) (美國Santa Cruz公司,批號:sc-173)。
1.2 方法
1.2.1 細胞培養 將人肝癌hepG2細胞株培養于含10%胎牛血清的RMPI-1640 培養液中,于37℃,5%CO2飽和濕度的孵箱中培養,待細胞貼壁生長后,2 d傳代1次,培養至對數生長期用于實驗。
1.2.2 MTT法檢測索拉菲尼對HepG2細胞增殖的影響 取對數生長期的HepG2細胞,以5×103/孔的密度接種于96 孔板中,培養24 h后加入藥物。實驗分成4組:索拉非尼(5、10、20 μmol/L)用藥組和不加藥的對照組。每個劑量設5個復孔,
應用SPSS17.0統計軟件進行數據處理,數據以()表示。計量資料多組間比較采用單因素方差分析,兩兩比較采用LSD-t法,P<0.05為差異有統計學意義。加藥后分別培養 12、24、48、72 h,再在每孔中加入 5 mg/mL的MTT 20 μL,繼續培養4 h后終止培養,棄去上清液,每孔中另加150μL DMSO,振蕩10 min,使結晶物充分溶解,最后用酶標儀測定各孔570 nm處的OD值,同時計算各組藥物對細胞增殖的抑制率,抑制率(%)=(1-實驗組OD 值/對照組OD 值)×100%。
1.2.3 各組細胞CDK5表達水平的檢測 取對數生長期的HepG2細胞,用0.25%胰蛋白酶消化后制成濃度為1×106個/mL的單細胞懸液,接種在蓋有玻片的6孔板中(2 mL/孔),24 h后細胞貼壁,棄去培養液;實驗組中分別加入濃度為5、10、20μmol/L含索拉菲尼的培養液,而對照組中加不含索拉菲尼的培養液(2 mL/孔)。將4組細胞在37℃、5%CO2孵箱中培養48 h后在倒置顯微鏡下觀察HepG2細胞的生長情況。拿出6孔板,分別用PBS洗滌3次(2min/次),之后用4%多聚甲醛室溫固定30 min(2 mL/孔)。采用免疫細胞化學染色法測定各組細胞CDK5的表達水平,實驗所有操作步驟均嚴格按試劑盒說明書進行。根據細胞著色的強弱程度反應CDK5的表達情況。
1.3 統計學處理
2 結果
2.1 索拉菲尼對人肝癌細胞HepG2 增殖的影響
MTT法結果顯示,索拉菲尼能明顯抑制人肝癌HepG2細胞的生長,且呈明顯的時間-劑量依賴效應。隨著用藥時間的延長,抑制率明顯上升; 隨著用藥濃度的提高,抑制率逐漸升高,各組間兩兩比較后差異均有統計學意義(P<0.05)。見表1、圖 1。

圖1 不同濃度索拉非尼作用不同時間對HepG2細胞的抑制作用
表1 MTT 法檢測不同濃度索拉菲尼對HepG2細胞增殖的抑制作用(OD 值,±s)
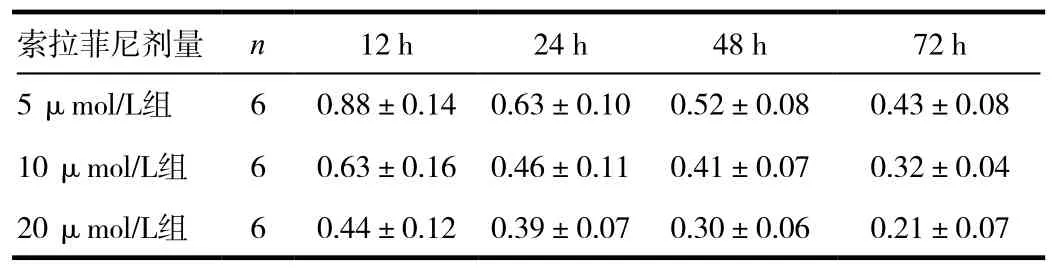
表1 MTT 法檢測不同濃度索拉菲尼對HepG2細胞增殖的抑制作用(OD 值,±s)
?
2.2 CDK5 在人肝癌細胞株HepG2中的表達
在倒置顯微鏡下觀察發現:實驗組的HepG2細胞形態已發生改變,較多的貼壁細胞由多形性變為透亮的圓形細胞,而且懸浮細胞的比例較對照組明顯增多(圖2)。光鏡下亦觀察到CDK5陽性表達產物呈棕黃色顆粒,在細胞核和細胞質中呈彌漫均質分布,且各組細胞陽性著色顆粒的大小、分布密度及著色深淺亦有不同,隨索拉菲尼的藥物濃度增大,胞核淡染的比例增大且著色逐漸變淡(圖3)。A:對照組;B:5 μmol/L組;C:10μmol/L組;D:20μmol/L組。
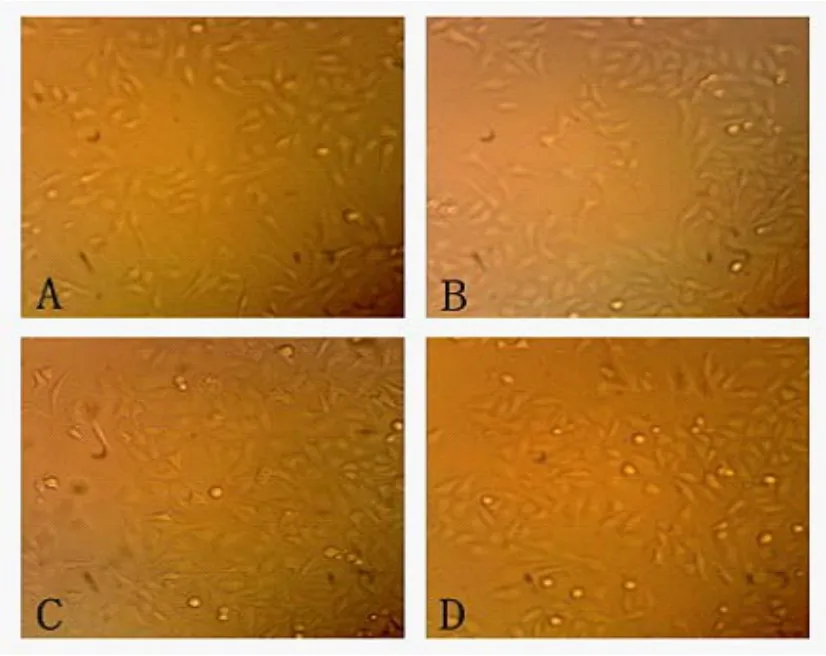
圖2 倒置顯微鏡下觀察
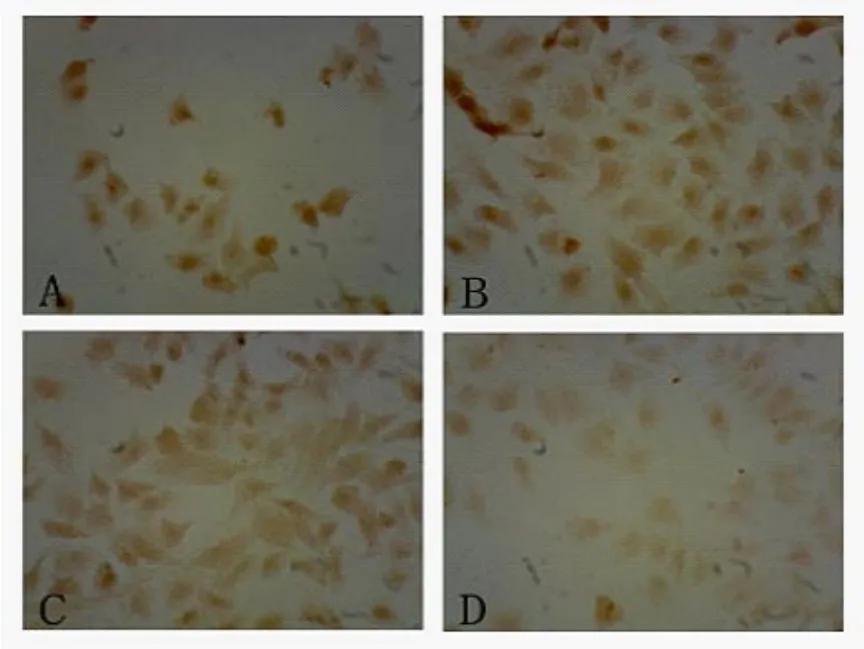
圖3 光鏡下觀察
3 討論
原發性肝癌的發生發展是一個多基因、多階段的發展過程,隨著分子生物學的不斷發展,人們逐漸認識到肝癌的發生發展是由一系列分子事件構成的,比如:原癌基因和抑癌基因的平衡失調、生長因子受體的失調、信號轉導的異常、血管生長因子以及基質金屬蛋白酶分泌的異常等[2]。而分子靶向藥物的飛速發展為治療原發性肝癌提供了新的選擇,并且新型分子靶向藥物也在臨床實踐中取得了顯著療效。原發性肝癌的分子靶向治療,主要是以腫瘤細胞過度表達的細胞受體、關鍵基因和某些標志性分子為靶點,通過選擇特異性阻斷劑對上述靶點和與其相關的信號通路進行有效的干預和調控,進而達到抑制腫瘤生長、發展及轉移的效果,具有特異性強、療效顯著、基本不損傷正常組織等優勢[3]。與其他惡性腫瘤一樣,肝癌中調控細胞生長的信號系統也處于失衡狀態,這樣其細胞則無限制生長并產生惡性表型。已有研究發現其主要信號通路有:Raf/MEK/ERK信號通路、Janus激酶信號轉導子及轉錄激活子通路、P13 K/Akt/mTOR信號通路、WNT/β-連環蛋白信號通路[4]和核因子кB等。其共同機制為將細胞外異常信號轉入細胞核內,通過調控細胞生長、分化和周期等導致肝癌的形成和發展。
索拉非尼是一種新型多靶點信號轉導抑制劑,一方面通過靶向作用于Raf/MEK/ERK信號轉導通路中的Raf激酶來阻斷肝癌細胞的增殖;另一方面通過抑制VEGFR-2/3受體激酶活性而發揮抗血管生成效應;亦有研究發現其還能誘導肝癌細胞凋亡[5-7]。臨床Ⅱ期及Ⅲ期實驗已表明索拉非尼對腎癌、肝癌、黑素瘤和非小細胞肺癌都有一定的治療作用。FDA已批準索拉非尼用于腎癌、肝癌的治療。索拉菲尼已被循證醫學證實可以顯著延長肝癌患者的中位生存時間[8-9]。
細胞周期素依賴性蛋白激酶(CDK)是細胞周期調控網絡的核心蛋白,其表達活性的改變將直接影響細胞周期的長短,這與機體細胞的生長、分化、凋亡以及腫瘤的發生發展及轉移密切相關[10-11]。CDK5作為CDK家族中的一員,有60%的序列與CDK2同源, 同時也包含了全部保守的蛋白激酶區域, 并且含有該家族成員共有的PSTAIRE結構域[12],但與其他成員不同,CDK5既非周期素依賴,也不參與細胞周期的調節,而是與p35、p39等調節亞基相互作用在有絲分裂后神經元復雜遷移、突觸傳遞和神經細胞死亡等活動中起重要作用。然而,最新研究已發現CDK5并不僅僅存在于中樞神經系統,在甲狀腺髓樣癌、肺癌、乳腺癌、前列腺癌等多種腫瘤組織中均有異常表達, 其主要通過影響細胞增殖及凋亡而導致腫瘤的發生發展。董賈中等[13]研究亦發現CDK5在肝細胞癌中也呈上調表達趨勢,且與腫瘤的分化程度相關。他們推測其原因可能為:(1)CDK5表達蛋白在肝癌細胞中發生了基因突變,導致CDK5蛋白結構表達異常,從而使CDK5蛋白產生跨膜運動,最終在細胞核內堆積;(2)CDK5作用于肝癌細胞內的轉錄因子MEF-2,導致其轉錄發生異常;(3) CDK5亦通過PIKE-A-Akt途徑活化Akt,進而激活mTOR信號傳導通路,最終引起腫瘤的發生發展;那么,如果應用CDK5的抑制劑則可以通過減少肝癌細胞核內CDK5/P35和磷酸化STAT3,進而延緩肝癌的發生發展。可見,CDK5的高表達在肝癌的發生發展過程中起著重要的作用,其可作為肝癌早期診斷的候選基因,但具體作用機制將有待于進一步研究。
本實驗結果顯示:索拉菲尼作用于肝癌細胞后,其增殖受到明顯的抑制,且CDK5的表達陽性率亦顯著降低。而CDK5表達水平下降可能的原因:一方面與阻斷Raf/MEK/ERK信號通路有關,在信號轉到過程中相關細胞因子發生改變,干擾相關信號轉導通路的順利進行,使得CDK5的表達下調;另一方面索拉菲尼本身可以誘導肝癌細胞凋亡,存活的肝癌細胞則減少,那么CDK5的合成水平則下降,其調節腫瘤發生發展的作用則減弱。可見,動態觀察CDK5的水平變化可作為判斷肝癌患者病情及療效的一項重要的指標。
然而,原發性肝癌細胞中信號轉導的過程是一個復雜的、多種因素相互交叉的蛋白網絡系統,若只切斷一個靶點進行治療顯然是不全面的。因此,必須尋找更多有效治療靶點,如通過多分子與多通路靶點藥物間的聯合治療、針對同一信息轉導通路的接收器及其下游轉導通路中任一階段的干預進行的合并治療以及不同分子靶向藥物之間互相聯合等,有待進一步探索。
[1] L lovet JM,B ruix J.Molecular targeted therapies in hepatocellular carcinoma[J].Hepatology,2008, 48(4):1313-1327.
[2] Villanueva A,Newell P,Chiang DY,et al.Genomlcs and signaling pathways in hepatoceilular carcinoma[J].Semin Liver Dis,2007,27(1):55-76.
[3] Vogelstein B,Kinzler KW.Cancer genes and the pathways theycontrol[J].Nat Med,2004,10(8):789-799.
[4] Thompson MD,Monga SP.WNT/beta-eatenin signaling in liver health and disease[J].Hepatology,2007,45(5):1298-1305.
[5] Flaherty KT.Sorafenib:delivering a targeted drug to the targets[J].Expert Rev Anticaneer Ther,2007,7(5):617-626.
[6] Hampton T.Cancer drug trials show modest benefit:drugs target liver,gastric,head and neck caners[J].JAMA,2007,298(3):273-275.
[7] Liu L,Cao YC,Chen C,et al.Sorafenib blocks the RAF/MEK/ERK pathway,inhibits tumor anglogenesis,and induces tumor cell apoptcsis in hepatocellular carcinoma model PLC/PRF/5[J].Cancer Res,2006,66(24):1851-1858.
[8] Lovet JM,Ricci S,Mazzaferro V,et al.Sorafenib in advanced hepatocellular carcinoma[J].NEngl J Med,2008,359(4):378-390.
[9] Cheng AL,Kang YK,Chen Z,et al.Efficacy and safety of sorafenib in patients in the AsiaPacific region with advanced hepatocellular carcinoma:a phaseⅢrandomisod,Double-blind,placebo-controlled trial[J].Lancet Oncol,2009,10(1):25-34.
[10] Liu L,Schwartz B,Tsubota Y,et al.Cyclindependent kinase inhibitors block leukocyteadhesion and migration[J].Immunol,2008,180:1808-1817.
[11] Cai D,Latham VM Jr,Zhang X,et al.Combined depletion of cell cycle and transcriptional cyclin-dependent kinase activities induces apoptosis in cancer cells[J].Cancer Res,2006,66:9270-9280
[12] Tarricone C,Dhavan R,Peng J,et al.Structure and regulation of the CDK5-p25(nck5a)complex[J].Mol Cell,2001,8:657-669
[13] 董賈中,楊玉秀, 白楊秋,等.CDK5在肝細胞癌中的異常表達[J].世界華人消化雜志,2010,18(10):1010-1015.
猜你喜歡
天津醫科大學學報(2019年3期)2019-08-13 06:53:08
中成藥(2016年8期)2016-05-17 06:08:14
癌癥進展(2016年12期)2016-03-20 13:16:17
罕少疾病雜志(2016年5期)2016-03-11 16:34:44
吉林大學學報(醫學版)(2015年1期)2015-12-17 07:47:28
腫瘤預防與治療(2015年1期)2015-09-26 07:26:20
中國當代醫藥(2015年16期)2015-03-01 02:03:11
中國醫藥導報(2015年26期)2015-02-28 22:07:59
肝膽胰外科雜志(2015年4期)2015-02-27 11:12:34
肝膽胰外科雜志(2015年4期)2015-02-27 11:12:24
