新型樹枝狀鎳系催化劑的合成、表征及催化乙烯聚合的性能
2014-07-05 16:04:25施偉光楊光王斯晗李翠勤王俊
化工進展 2014年7期
關鍵詞:催化劑
施偉光,楊光,王斯晗,李翠勤,王俊
(1東北石油大學化學化工學院石油與天然氣化工省重點實驗室,黑龍江 大慶 163318;2中國石油大慶化工研究中心,黑龍江 大慶 163318)
新型樹枝狀鎳系催化劑的合成、表征及催化乙烯聚合的性能
施偉光1,楊光1,王斯晗2,李翠勤1,王俊1
(1東北石油大學化學化工學院石油與天然氣化工省重點實驗室,黑龍江 大慶 163318;2中國石油大慶化工研究中心,黑龍江 大慶 163318)
合成了一種含有強供電子、較大空間位阻的新型樹枝狀水楊醛亞胺配體及其鎳配合物,利用1H NMR、元素分析、FT-IR、UV進行了表征。以配合物為催化劑,甲基鋁氧烷(MAO)為助催化劑對乙烯齊聚反應進行了研究,詳細考察了聚合反應溫度、Al/Ni摩爾比、反應壓力及時間對催化劑活性及產物分布的影響。結果表明,齊聚產物為不同碳鏈的烯烴,在溫度為25℃、Al/Ni=500、壓力為0.5MPa、時間為0.5h的條件下,樹枝狀催化劑的活性最高,達到4.93×105g/(mol Ni·h),C10~C18的含量為54.17%。
樹枝狀大分子;水楊醛亞胺配體;鎳配合物;乙烯齊聚
樹枝狀大分子的合成與應用已有近三十年的歷史,大量的樹枝狀大分子被開發出來,這類具有獨特結構的有機高分子在催化領域展示出良好的前景,對這類分子的催化作用研究幾乎涉及所有的有機合成反應[1-2]。近年來,樹枝狀金屬配合物作為一類新型的催化劑在烯烴聚合領域取得了重大的研究進展[3-4],尤其是通過對樹枝狀金屬配合物的修飾,設計出具有不同性能的烯烴聚合催化劑,達到調變聚合產物分布的作用[5-6]。本文作者課題組合成了用于烯烴聚合反應的樹枝狀水楊醛亞胺鎳配合物[7],在此基礎上設計合成了含有強供電子、較大空間位阻的樹枝狀水楊醛亞胺配體及其鎳配合物,以甲基鋁氧烷(MAO)為助催化劑催化乙烯聚合反應并考察了聚合條件對催化性能及產物分布的影響。
1 實驗部分
1.1 試劑與儀器
配體及配合物在氮氣氛圍下應用標準Slchenk技術進行合成;3,5-二叔丁基水楊醛,分析純,湖北康寶泰精細化工有限公司;無水氯化鎳,分析純,Aldrich公司;MAO,10%的甲苯溶液,Aldrich公司;聚合級乙烯和高純氮氣,大慶石化公司;甲苯及甲醇均為分析純,天津市津北精細化工有限公司,使用前脫水脫氧處理并在氮氣條件下回流蒸出;1.0代乙二胺為核的聚酰胺-胺(1.0G PAMAM),自制[7]。
CHN-O Rapid型元素分析儀,德國Heraeus;Bruker-Vector 22型傅里葉變換紅外光譜儀;INOVA 400MHz型核磁共振儀,美國Varian公司;UV-1700 PharmaSpec型紫外可見分光光度計,深圳市科美嘉儀器設備有限公司;HP-5890氣相色譜儀。
1.2 樹枝狀水楊醛取代物亞胺配體的合成
將2.3g(4.46mmol)1.0G PAMAM(Ⅰ)溶解于80mL無水乙醇中,加入到帶有磁力攪拌子和回流冷凝管的250mL三口圓底燒瓶中,同時加入3.0g無水硫酸鈉,氮氣保護下78℃回流攪拌10min后,將8.8g(37.61mmol)3,5-二叔丁基水楊醛(Ⅱ)溶解于20mL乙醇中,采用恒壓滴液漏斗將其緩慢滴加到三口圓底燒瓶中,氮氣保護下反應12h后趁熱抽濾,濾液放置于冰箱中靜止36h,待亮黃色晶體析出后,抽濾并將晶體用無水乙醇進行洗滌、重結晶,50℃真空烘干,即得樹枝狀水楊醛亞胺配體(Ⅲ),合成路線見圖1。產率為78.79%。
1.3 樹枝狀金屬配合物的合成
取配體Ⅲ 1.1868g (0.86mmol)溶于18mL無水甲醇,加入到帶有磁力攪拌子的茄形瓶中,在氮氣保護下25℃攪拌10min;采用恒壓滴液漏斗緩慢滴加用18mL無水甲醇溶解的氯化鎳(Ⅳ) 0.1276g(0.98mmol)溶液,反應24h后立即將反應混合物放入冰浴中,抽濾并用少量無水甲醇進行洗滌;將所得晶體在30℃真空下烘干,即得樹枝狀橋聯鎳配合物(Ⅴ),合成路線見圖2。產率為76.35%。
1.4 乙烯聚合反應
所有聚合反應均在無水、無氧的條件下進行。將帶有磁攪拌子的250mL聚合反應釜加熱抽真空并用氮氣連續置換3次,在氮氣氛下,加入50mL甲苯溶液,再通入乙烯,使乙烯充分吸收至飽和,然后用注射器將助催化劑MAO加入反應瓶中,攪拌15min后注入1mL催化劑(5mg/mL的甲苯溶液),通過調節乙烯通入量控制聚合體系壓力。25℃、0.5MPa、反應0.5h、Al/Ni比為500的條件下,保持其中3個條件恒定,考察另一個反應條件對催化劑聚合活性及產物分布的影響。反應結束后,放空乙烯氣體,稱重,計算活性,并取1μL反應混合物進行GC分析,然后用100mL 2%鹽酸-甲醇溶液終止反應。

圖1 樹枝狀水楊醛亞胺配體(Ⅲ)合成路線
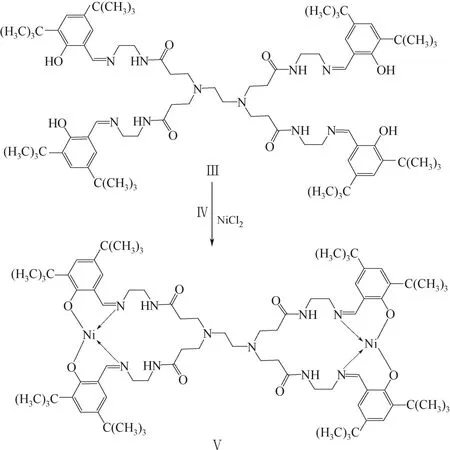
圖2 樹枝狀水楊醛亞胺配合物(Ⅴ)合成路線
2 結果與討論
2.1 配體及配合物的合成及表征
2.1.1 配體及配合物的元素分析
含有強供電子、較大空間位阻的樹枝狀水楊醛亞胺配體及其鎳配合物的元素分析結果見表1。數據表明,配體及配合物元素的理論值與計算值基本一致。
2.1.2 配體及配合物的紅外譜圖分析
紅外光譜檢測得到光譜如圖3所示,圖中C為樹枝狀金屬配合物,L為樹枝狀亞胺配體。
配體L在1628cm-1處出現了C=N的伸縮振動吸收峰,表明1.0G PAMAM與水楊醛已經發生了席夫堿反應[7]。由于金屬與氮原子之間發生了配位,削弱了C=N雙鍵的強度,加強了碳原子的C—H鍵彎曲振動,從而使ν(C=N) 吸收峰移動至 1622 cm-1處。2993cm-1及2998cm-1為配體及配合物中ν(CONH)的特征吸收峰。配體中—OH在3409cm-1處出現伸縮吸收峰,配位后ν(OH)在3409cm-1附近的吸收峰消失,表明—OH失去氫原子參與配位。Ν(Ar—O)伸縮振動由1041cm-1移動到1089cm-1,也表明—OH參與配位。同時,配體及配合物中在2990cm-1、3400cm-1附近都出現了波數偏小的酰胺N—H鍵伸縮振動峰,這是由于N—H之間形成氫鍵,使波數偏小。
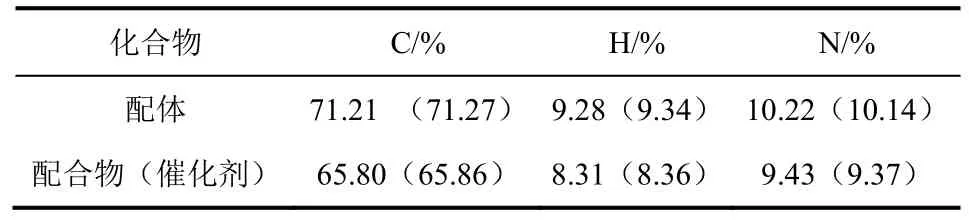
表1 樹枝狀配體及其鎳配合物的元素分析
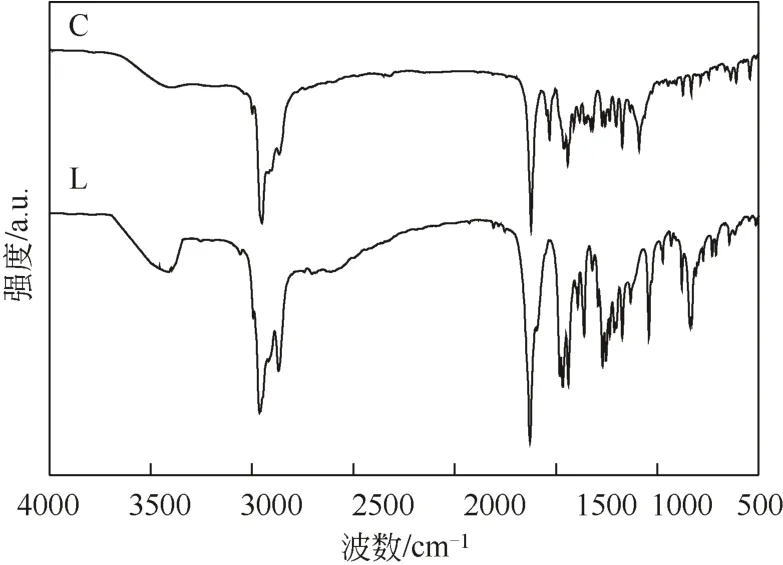
圖3 樹枝狀亞胺配體及金屬配合物紅外譜圖
2.1.3 配體及配合物的紫外分析
以乙醇為溶劑,樹枝狀水楊醛亞胺配體及其鎳配合物的紫外光譜數據如表2所示。紫外可見光譜中的200~242nm、244~289nm、289~381nm的吸收為配體內躍遷。在樹枝狀配合物中,3個位置發生明顯的變化。在配體中,223nm、264nm強吸收峰是苯環的E2帶及苯環與C=N基共軛的K帶,苯環的B帶被K帶所掩蓋,而331nm處出現了的n→π*躍遷的R帶。當配體與金屬配位后,R帶紅移至363nm處,說明N原子參與了配位。同時N原子參與配位后,共軛作用減小,導致吸收譜峰發生了藍移,E2帶、K帶均向低波長方向移動。

表2 樹枝狀配體及樹枝狀催化劑的紫外可見光譜
2.1.4 配體及配合物的核磁分析
圖4為樹枝狀水楊醛亞胺配體結構。由于鎳配合物具有順磁性,無法進行1H NMR分析,配體的核磁分析結果如圖5。配體分子中H原子的化學位移如表3,其中3.10~4.00出現的質子峰為配體骨架中亞甲基上的特征質子峰。配體分子中在δ1.33和δ1.46處出現兩個甲基的特征質子峰,表明配體中存在叔丁基,即1.0G PAMAM與3,5-二叔丁基水楊醛發生了希夫堿反應。由于苯環上取代基—C=N— 和—OH對Hj、Hl的共同影響,使苯環上相對于—C=N—的鄰對位電子云密度降低,δHl值增大,間位電子云密度增加,δHj值減小[8]。當體系中存在多種活潑氫時,如樣品分子中既含有羧基,也含有羥基、胺基時,在他們均進行快交換的條件下,其核磁譜圖也只顯示一個綜合的、平均的活潑氫信號[9]。由于樣品吸收了水,故與配體中羥基、酰胺基中的活潑氫發生快速交換,在譜圖中只在δ4.87處顯示一個綜合的活潑氫信號。

圖4 樹枝狀水楊醛亞胺配體結構
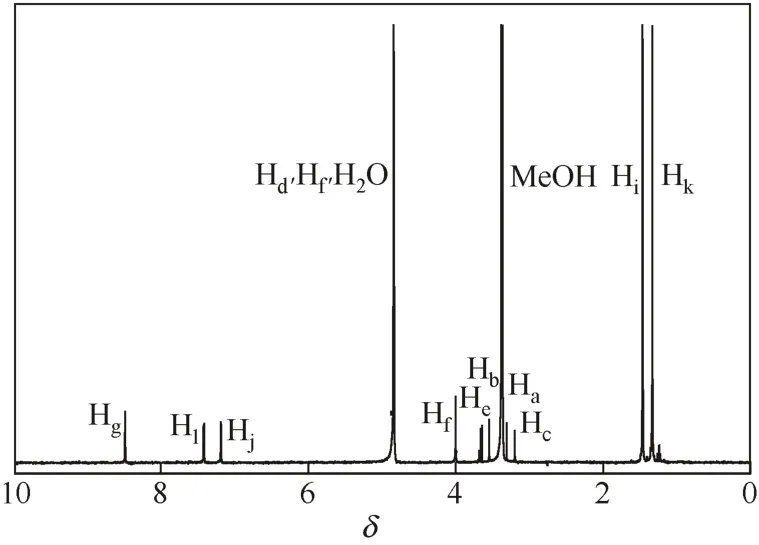
圖5 樹枝狀水楊醛亞胺配體的1H NMR譜圖
2.2 配合物/MAO體系催化性能研究
2.2.1 Al/Ni摩爾比對催化體系的影響
在催化反應體系中,MAO不但可以清除有害雜質和活化催化劑,還可以在烯烴聚合時充當鏈轉移劑[10]。如表4所示,隨著Al/Ni摩爾比的增加,催化劑的活性呈先增大后減小的趨勢。當Al/Ni摩爾比從100增加到500時,催化劑的活性增加幅度較大。這是由于在反應中Al/Ni摩爾比較低時,助催化劑MAO的量不足以完全除去體系中的有害雜質并將活性中心活化,隨著助催化劑量的增加,活性中心的數目增加,更利于催化活性的提高。當Al/Ni摩爾比大于500時,過多的MAO與活性中心反應導致活性中心失活,故催化活性隨Al/Ni摩爾比的增加而降低。所以對該催化劑來說,Al/Ni摩爾比為500時表現出最佳催化活性。以上實驗結果與Chen等[11]報道的烯烴聚合催化劑的結果吻合。由于鏈增長的速率始終高于鏈轉移的速率,因此GC測試的結果顯示,聚合產物主要由C10~C18的烯烴組成,在Al/Ni摩爾比為100時,C10~C18的含量達到最高,為76.70%。

表3 樹枝狀水楊醛亞胺配體中活潑氫的化學位移
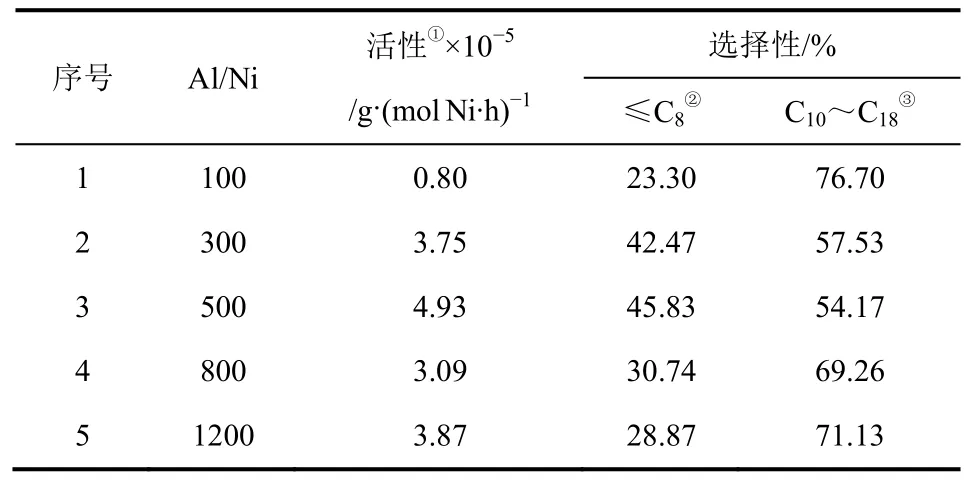
表4 Al/Ni摩爾比對配合物催化活性及聚合產物分布的影響
2.2.2 聚合反應時間對催化體系的影響
反應時間對樹枝狀水楊醛亞胺鎳系催化劑催化性能的影響如表5所示。在反應開始階段,催化劑的活性最大,隨著反應時間的增加,催化活性下降,這說明乙烯齊聚反應主要發生在開始階段。另一方面,較長的反應時間使催化活性中心失活[12]。延長反應時間有利于鏈增長過程,在反應時間為100min時,C10~C18含量高達65.83%。
2.2.3 聚合反應壓力對催化體系的影響
如表6所示,隨著聚合壓力的增大,催化反應活性不斷增加,這是由于隨著聚合壓力的增大,乙烯氣體在溶劑甲苯中的溶解度增大。因此在0.5MPa時,催化劑活性達到最高值4.93×105g/(mol Ni·h),高于Smith等[13]報道的樹枝狀烯烴聚合催化劑的活性。從表6中可以看出,隨著聚合壓力的增大,碳數分布向碳數增大的方向移動,即C10~C18的含量增多。因為隨著聚合壓力的提高,抑制了β-H消除反應的速率而增加了鏈增長速率,從而有利于形成高碳烯烴[14-15]。
2.2.4 聚合反應溫度對催化體系的影響
反應溫度對樹枝狀水楊醛亞胺鎳系催化劑的催化活性和選擇性的影響見表7。隨著聚合反應溫度的升高,催化活性逐漸升高,在35℃時達到最大值6.13×105g/(mol Ni·h),而后隨著溫度的升高催化活性開始降低。在溫度較低時,催化劑的活性中心不容易被活化,使得活化中心的數量少,同時鏈增長速率小,且乙烯在溶劑甲苯中的溶解度小,故低溫時催化活性低;隨著溫度的升高,乙烯在甲苯中的溶解度不斷減小,且越來越多的活性中心被MAO活化,鏈增長速率增加,催化活性逐漸升高并在35℃時達到最高值。但在高溫時會導致催化劑失活,故聚合速率降低[16-17]。此外,隨著反應溫度的變化,鏈增長速率并不是始終高于鏈轉移速率,因此聚合產物中C10~C18的含量隨溫度變化無明顯規律。

表5 反應時間對配合物催化活性及聚合產物分布的影響
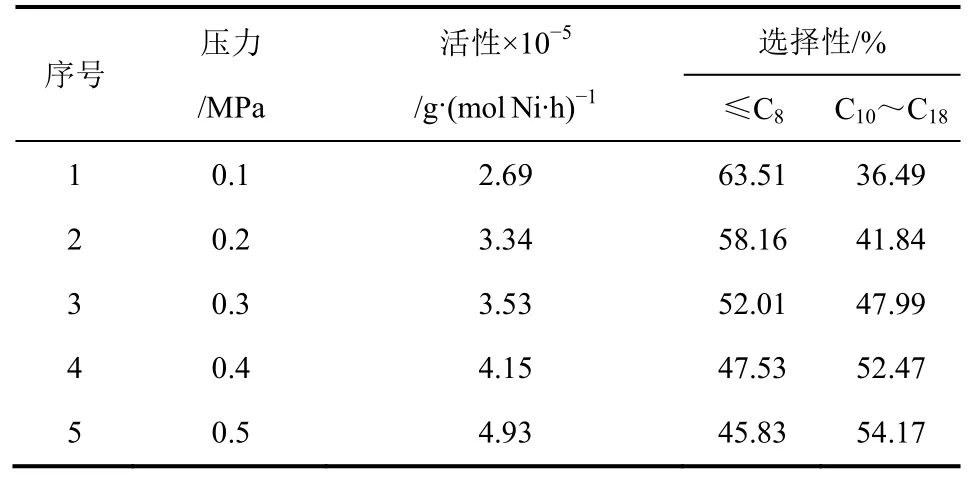
表6 反應壓力對配合物催化活性及聚合產物分布的影響
3 結 論
(1)以1.0G PAMAM、3,5-二叔丁基水楊醛、無水氯化鎳為原料,合成了一種含有強供電子、較大空間位阻的樹枝狀水楊醛亞胺配體及其相應的鎳配合物。通過一系列的表征實驗證明所合成的配體及配合物的結構與理論結構相符。
(2)在以MAO為助催化劑的作用下,樹枝狀水楊醛亞胺鎳配合物同樣可催化乙烯聚合反應,這種催化劑與同類催化劑相比活性相當,但這種含有強供電子、較大空間位阻的催化劑對聚合產物的分布有明顯的影響。在25℃、0.5MPa、Al/Ni=500、0.5h的聚合反應條件下,催化劑的活性最高達4.93×105g/(mol Ni·h),產物中C10~C18的含量
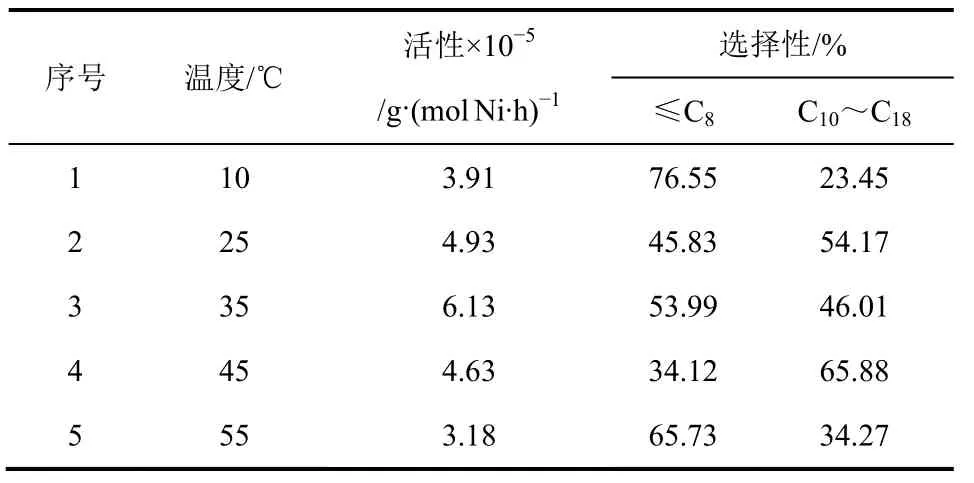
表7 反應溫度對配合物催化活性及聚合產物分布的影響
為54.17%。
[1] Newkome G R,Yao Z,Baker G R. Chemistry of micelles series. Part 2. Cascade molecules. Synthesis and characterization of a benzene [9]3-arborol[J].Journal of the American Chemical Society,1986,108(4):849-850.
[2] Svenson S,Tomalia D A. Dendrimers in biomedical applicationsreflections on the field[J].Advanced Drug Delivery Reviews,2012,64:102-115.
[3] Malgas-Enus R,Mapolie S F. Nickel metallodendrimers as catalyst precursors in the tandem oligomerization of ethylene and Friedel-Crafts alkylation of its olefinic products[J].Inorg. Anica. Chimica. Acta.,2014,409(1):96-105.
[4] 王俊,王海琛,李翠勤,等. 樹枝狀烯烴聚合催化劑研究進展[J]. 石油學報:石油加工,2013,29(5):920-928.
[5] Benito J M,Jesús E,Mata F J,et al. Mononuclear and dendritic nickel(Ⅱ) complexes containingN,N'-iminopyridine chelating ligands:Generation effects on the catalytic oligomerization and polymerization of ethylene[J].Organometallics,2006,25(16):3876-3887.
[6] Koten G V,Jastrzebski J T B H. Periphery-functionalized organometallic dendrimers for homogeneous catalysis[J].Journal of Molecular Catalysis A:Chemical,1999,146:317-323.
[7] Wang J,Zhang P,Chen S,et al. The preparation of dendritic nickel complex and performance evaluation in the oligomerization of ethylene[J].Journal of Macromolecular Science,Part A:Pure andApplied Chemistry,2013,50(2):163-167.
[8] 陳潔,宋啟澤. 有機波譜分析[M]. 北京:北京理工大學出版社,1996:101-112.
[9] 寧永成. 有機化合物結構鑒定與有機波普學[M]. 北京:科學出版社,2000.
[10] McGuinness D S,Rucklidge A J,Tooze R P,et al. Cocatalyst influence in selective oligomerization: Effect on activity,catalyst stability,and 1-hexene/1-octene selectivity in the ethylene trimerization and tetramerization reaction[J].Organometallics,2007,26(18):4696-4696.
[11] Chen J X,Huang Y B,Li Z S,et al. Syntheses of iron,cobalt,chromium,copper and zinc complexes with bulky bis(imino)pyridyl ligands and their catalytic behaviors in ethylene polymerization and vinyl polymerization of norbornene[J].Journal of Molecular Catalysis A:Chemical,2006,259(1-2):133-141.
[12] Doherty M D,Trudeau S,White P S,et al. Ethylene oligomerization catalyzed by a unique phosphine-oxazoline palladium(Ⅱ) complex. Propagation and chain transfer mechanisms[J].Organometallics,2007,26(5):1261-1269.
[13] Smith G,Chen R,Mapolie S. The synthesis and catalytic activity of a first-generation poly(propylene imine) pyridylimine palladium metallodendrimer[J].Journal of Organometallic Chemistry,2003,673(1-2):111-115.
[14] Malgas R,Mapolie S F,Ojwach S,et al. The application of novel dendritic nickel catalysts in the oligomerization of ethylene[J].Catalysis Communications,2008,9(7):1612-1617.
[15] 羅勤慧. 配位化學[M]. 北京:科學出版社,2012:407-413.
[16] Mogorosi M M,Mahamo T,Moss J R,et al. Neutral palladium(II)complexes with P,N Schiff-base ligands:Synthesis,characterization and catalytic oligomerisation of ethylene[J].Journal of Organometallic Chemistry,2011,696(23):3585-3582.
[17] Ojwach S O,Guzei I A,Benade L L,et al. (Pyrazol-1-ylmethyl)pyridine nickel complexes:Ethylene oligomerization and unusual friedel-crafts alkylation catalysts[J].Organometallics,2009,28(7):2127-2133.
Synthesis and characterization of novel dendrimer-based nickel catalyst for ethylene polymerization
SHI Weiguang1,YANG Guang1,WANG Sihan2,LI Cuiqin1,WANG Jun1
(1Provincial Key Laboratory of Oil & Gas Chemical Technology,School of Chemistry & Chemical Engineering,Northeast Petroleum University,Daqing 163318,Heilongjiang,China;2Daqing Petrochemical Research Center,CNPC,Daqing 163318,Heilongjiang,China)
A newtert-butyl-substituted dendrimer compound and its corresponding Ni(Ⅱ) complex were synthesized and characterized with1H NMR,elemental analysis,FT-IR,UV. The nickel complex activated by methylaluminoxane (MAO) was tested in oligomerization of ethylene. The effects of Al/Ni molar ratio and polymerization temperature on catalyst activity and product distribution were also investigated. The products were olefines with different carbon chains,catalytic activity reached a maximum value 4.93×105g/(mol Ni·h) at reaction time of 30min,reaction pressure of 0.5MPa,reaction temperature of 25℃ and Al/Ni ratio 500 for thetert-butyl-substituted nickel complex,and C10—C18were 54.17%.
dendrimer;salicylaldehyde-imine ligand;nickel complex;ethylene oligomerization
O 643.3
A
1000-6613(2014)07-1763-06
10.3969/j.issn.1000-6613.2014.06.018
2014-01-16;修改稿日期:2014-02-26。
施偉光(1979—),男,講師,碩士生導師。聯系人:李翠勤,副教授,主要從事新型表面活性劑及合成材料助劑的結構與性能研究。E-mail licuiqin78@163.com。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
