過渡金屬合金催化劑催化作用機理研究進展
2014-08-08 09:52:38崔鑫林瑞趙天天楊美妮馬建新
化工進展 2014年1期
關鍵詞:催化劑
崔鑫,林瑞,趙天天,楊美妮,馬建新
(1同濟大學新能源汽車工程中心,上海201804;2同濟大學汽車學院,上海201804)
質子交換膜燃料電池(PEMFC)能直接將化學能轉換為電能,相比于傳統內燃機,具有無污染的優點;相比于風能、太陽能、地熱能、核能等新型能源,具有能量和功率密度高的優點;相比于高溫燃料電池,具有啟動快、結構簡單的優點,被公認為是電動汽車、固定發電站等的首選能源,也是目前應用最為廣泛的燃料電池[1]。燃料電池所用電催化劑以鉑為主,活性高,催化性能好,但鉑資源稀少,價格昂貴,是阻礙PEMFC商業化的主要限制因素。因此,降低貴金屬用量、提高催化劑的活性是催化劑研究的主要目的。
氫電極交換電流密度約為10-3A/cm2,可逆性高,氧電極的交換電流密度僅為10-9A/cm2,電化學極化高[2]。陰極氧還原的標準可逆電極電勢為1.23V,而實際工作的電勢低于0.8V,電勢損失是陽極氫氧化反應的10倍,
如不使用催化劑,會嚴重影響PEMFC的電壓效率及電池輸出功率[3]。因此提高陰極催化劑的氧還原活性及穩定性、減少Pt的用量、降低成本是PEMFC電催化劑研究的主要目標。
降低Pt的用量、提高陰極電催化劑的性能,可以通過改變催化劑載體[4-5]、優化制備工藝和催化劑合金化[6]三種方法實現。近些年來,對Pt與其他貴金屬的合金催化劑已經有大量研究[7-8],其催化性能比純Pt提高很多。考慮到貴金屬稀少,Pt與過渡非貴金屬合金對燃料電池催化材料的發展具有重要的意義[9]。本文主要針對近幾年Pt-M(非貴金屬)合金催化劑的陰極氧還原機理和合金催化劑活性提高機理進行規律總結,綜述了添加的合金成分和合金結構對催化劑性能的影響。
1 氧還原反應機理研究進展
氧還原反應(ORR)為多電子反應,反應機理比氫還原反應復雜,涉及多步基元反應和不同中間產物(如O2-2、O-2、HO2-、H2O2及表面Pt-O和Pt-OH)的反應。對氧還原機理的研究需了解涉及的每步基元反應,生成的所有中間產物和反應動力學參數等。可能的反應途徑如圖1所示。
(1)直接反應途徑 直接發生4電子反應(速率常數為k1)生成水,如式(1)。
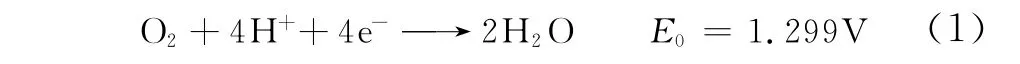
(2)連續反應途徑 先2電子反應生成中間產物H2O2,H2O2可能繼續發生2電子反應(速率常數為k3)還原生成H2O,也可能直接從溶液中析出生成產物H2O2,中間產物不穩定,還可能發生可逆反應分解為O2重新參加還原反應,如式(2)、式(3)。
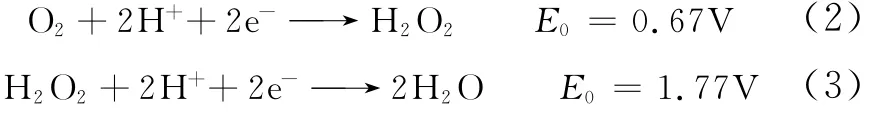
理想的燃料電池氧還原反應氧氣充分還原,輸出電壓高,為4電子反應途徑,由式(1)和式(2)知,發生4電子反應的還原電位比2電子反應高0.56V,而O2中O—O鍵的解離能498kJ/mol比H2O2中O—O鍵的解離能98.7kJ/mol大[11],因此催化活性不是很強時容易發生2電子反應或是2電子與4電子的混合反應。2電子反應會產生H2O2,對催化劑及質子交換膜造成損害,加速催化劑和質子交換膜的老化[12],因此4電子反應是理想的氧還原途徑。
O2在過渡金屬上的吸附模式大致分為Griftiths模式、Pauling模式和Birdge模式,如圖2所示。Griftiths模式中氧分子的軌道與中心原子空位的d2z軌道或dxz、dyz軌道有較強的相互作用,減弱O—O鍵,有利于O2發生直接4電子反應。Pauling模式中氧分子只有一側與過渡金屬作用,不利于O—O鍵的斷裂,一般發生2電子反應。Birdge模式中氧分子同時被兩個中心原子活化,有利于氧的4電子反應。
O2在催化劑表面的反應機理探究多以基元反應為基礎,來確定不同催化劑下反應速率的控制步驟。主要分為兩大方向對其進行研究:一為電子和質子轉移與化學鍵斷裂和生成;二為催化劑與中間產物的吸附關系。
1.1 電子和質子的轉移
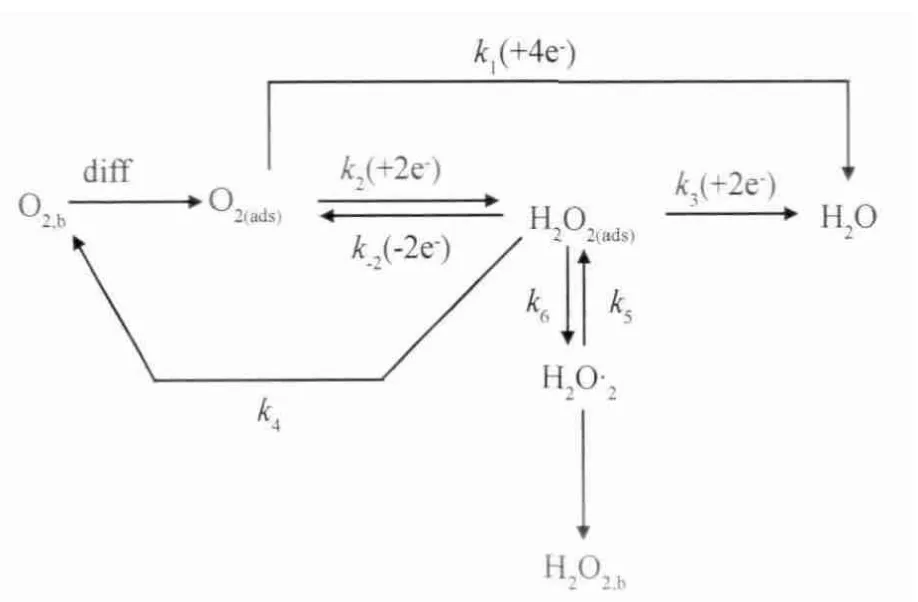
圖1 氧還原反應機理[10]
圖2 氧氣在過渡金屬表面的吸附模式
在電子和質子的轉移方面,很多研究者通過理論計算來分析氧還原反應的控速步驟。N?rskov等[13]在熱力學的基礎上,通過密度泛函理論對不同基元反應的吉布斯自由能進行計算,研究結果發現,反應途徑主要分為分離機理和聯合機理。其中,分離機理為O2分子斷開O—O鍵變成O原子,然后再進一步和質子電子反應生成H2O,其氧氣的解離為反應的控速步驟;而聯合機理中,O2分子直接結合電子變成,然后與質子反應生成,再進一步反應生成水,第一步的電子和質子的轉移為反應的控速步驟。該理論能對ORR的各基元反應的Gibbs自由能進行分析,但與真實的機理動力學仍存在一定的差異。Kalberg等[14]通過進一步的研究發現,當在電極附近施加一定的電場時,O2更容易發生聯合機理,即認為第一步電子和質子的轉移為反應的控速步驟。Hyman等[15]同樣采用密度泛函理論(DFT),用模型模擬酸性電解質溶液,研究了Pt(111)上的氧還原反應,認為O原子的質子化(即聯合機理)是反應的速率控制步驟,且生成的OH會阻礙O2反應,使O2在催化劑表面聚集。而Wakisaka等[16]通過EC-XPS分析了純Pt和Pt-Fe合金的氧還原反應,發現電極表面吸附的氧氣分子易發生O—O鍵斷裂變成游離態的氧原子,即認為分離機理為反應速率的控制步驟。
1.2 催化劑與中間產物的吸附
對于催化劑與中間產物的吸附關系,早在1913年,Paul Sabatier就提出了Sabatier原理,即認為催化劑對吸附的反應物、中間體和產物的吸附強度既不能太強,也不能太弱。如果吸附能力太強,就會導致催化劑的活性位點始終被占據,不利于新的反應物吸附,阻斷催化反應進行;如果作用太弱,則反應物不能很好地吸附在催化劑上,不利于催化反應發生。Sabatier原理是關于反應速率與吸附能的關系,而反應速率在一定程度上決定催化劑的活性。d帶中心理論認為金屬催化劑對物種的吸附強度與其表面原子d帶中心值呈線性關系。Lima等[17]采用d帶中心理論研究濃度為0.1mol/L的NaOH溶液中Pt/C、Au/C、Pd/C、Rh/C等貴金 屬 及Pt-Ru(0001)、Pt-Ir(111)、Pt-Rh(111)、Pt-Au(111)等合金的d帶中心位置和反應電流密度的關系,發現催化劑的催化活性與金屬的d帶中心的位置呈火山關系[18]。從圖3可以看出,對于貴金屬,只有當催化劑金屬的d帶中心(即催化劑對中間物種的吸附強度)在合適的范圍內,催化劑的催化活性才能達到最高,過大或過小的吸附強度都不利于催化反應的進行。如圖4所示,Stamenkovic等[19]研 究 了Pt3Fe、Pt3Co、Pt3Ti和Pt3Ni合金的d帶中心與催化性能的關系,發現過渡非貴金屬催化劑的活性同樣與d帶中心亦呈火山關系。金屬吸附O原子的能力太強,會造成催化劑活性表面始終被O原子占據,不利于催化反應持續進行;相反,如果金屬對O原子的吸附能力太弱,則會導致O2無法吸附在催化劑活性表面,不利于催化反應的發生。
2 Pt-M合金催化劑活性提高機理
燃料電池陰極催化劑主要以Pt/C為主,為提高Pt的催化活性,降低Pt用量,國內外學者通過各種物理(如真空濺射、離子噴涂等)和化學方法(沉積-還原、微乳、微波法等)將Pt分散到具有3d結構的過渡非貴金屬中,形成Pt-M二元、三元合金催化劑以提高氧還原反應的活性。Pt-M合金催化劑催化活性的提高機理主要分為以下幾個方面[20]。
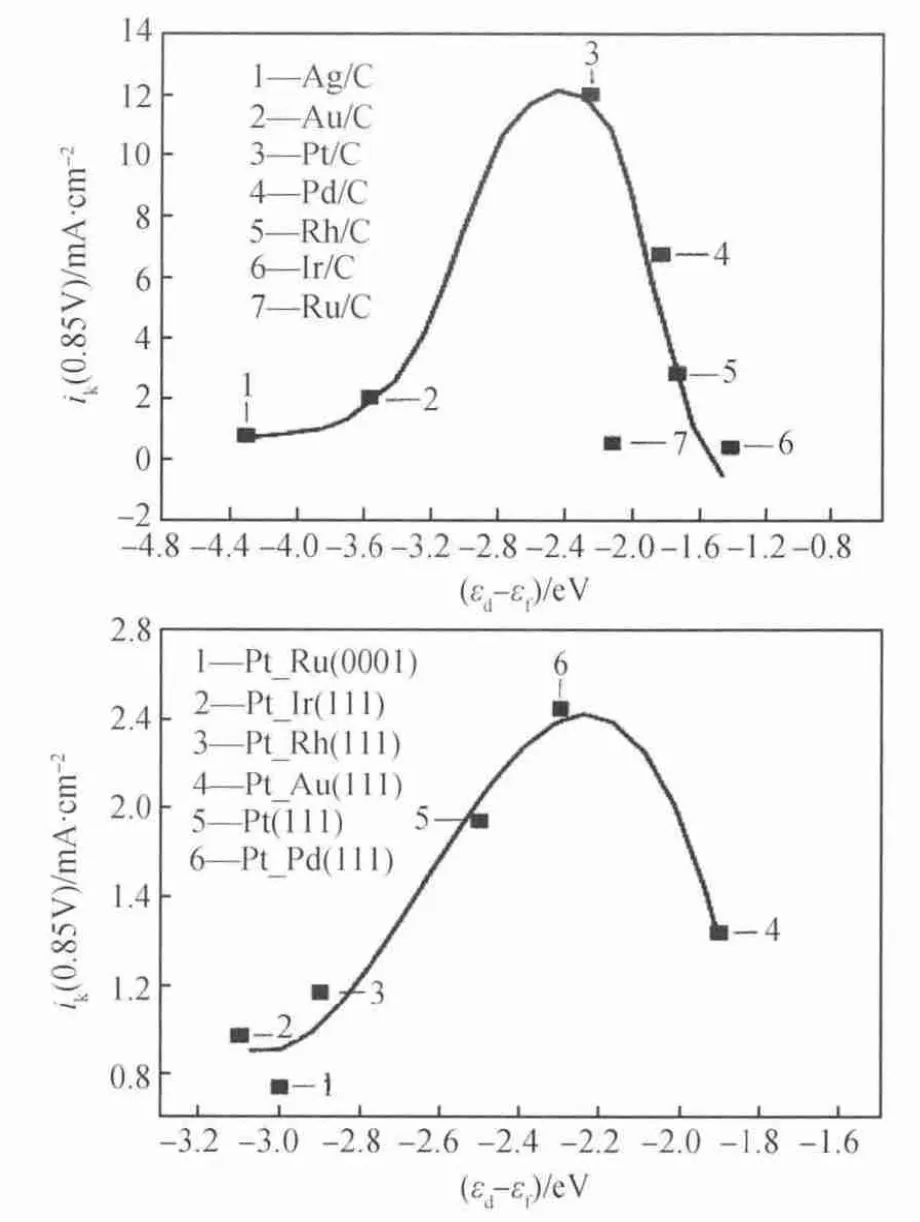
圖3 不同金屬的d帶中心與電流密度的關系[18]
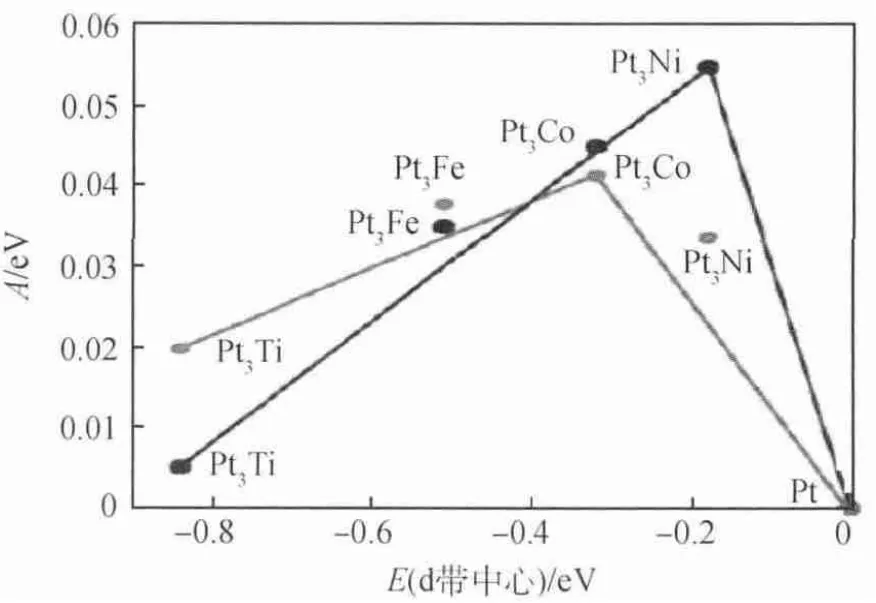
圖4 Pt3M(Ti、Fe、Ni、Co)的d帶中心與電流密度的關系[19]
2.1 幾何效應(最小原子間距理論)
O2在催化劑表面的Birdge吸附模式為雙位吸附,有利于O2進行4電子反應,產生更高的電壓,提高催化劑的活性。O2的雙位吸附與催化劑表面的活性結構密切相關,只有當O—O鍵的鍵長與催化劑兩個活性位點的距離相近時,O2才容易發生雙位吸附,被活化為O原子,進而與H+反應生成H2O。有學者認為由于非貴金屬M的添加使Pt-Pt的晶格間距收縮,Pt原子結構發生改變更有利于O2解離吸附,從而提高Pt-M催化劑的活性,即為幾何效應。Min等[21]采用初濕含浸法制備了二元鉑系合金Pt-Co、Pt-Cr、Pt-Ni,實驗測得Pt-M合金比純Pt催化劑的晶格參數更小,通過XRD和EXAFS分析發現合金的Pt-Pt原子間距變小。從圖5可以看出,合金的比活性隨著Pt-Pt原子間距的縮小而提高,其中Pt-Ni合金的比活性最高,且Pt-Pt原子間距最短。原因在于Pt-Pt原子間距的縮短有利于O2吸附解離,故表現出更高的催化活性。Kristian等[22]采用還原法制備了Co@Pt/C合金催化劑,氧還原活性是Pt/C催化劑的2~4倍。研究發現,Pt-Pt的原子間距和表面缺陷少是催化劑活性提高的原因,抑制了反應過程中Pt—OH的形成。
2.2 電子效應
合金元素的添加改變了Pt原子外層的電子結構,增大了Pt原子d軌道空穴數,增強了Pt原子d2z或兩個相鄰的Pt原子dxz或dyz軌道與吸附的O2分子π軌道的作用,降低了O—O鍵的鍵能,加快了O—O鍵斷裂,促進了氧還原反應的發生。Stamenkovic等[19]通過密度泛函理論對Pt及其合金的d帶中心與催化活性的關系進行預測,并與實驗測量數據進行比較,如圖4所示,發現預測活性最高點是Pt3Ni合金,Pt3Co合金次之,而實際測得Pt3Co合金催化活性最高,Pt3Ni次之,與理論計算結果基本一致。由d帶中心理論可知,O原子和金屬的吸附強度決定合金催化劑的活性,d帶中心的位置跟費米能級有關。我們可以通過改變d能帶中心的位置來控制合金間的耦合能,改善合金催化劑的活性。當d能帶中心上移(即d能帶中心能量變大),Pt-M與O2的相互作用增強,易于斷O—O鍵。反之,Pt-M上吸附的O原子易于和H結合生成OH。d帶中心理論僅僅是對合金催化活性的定性研究,定量研究還需復雜精確的理論計算。
2.3 雷尼效應(表面粗糙效應)
Pt-M合金催化劑中添加的非貴金屬溶解會使催化劑的表面變得粗糙,增加Pt的有效活性表面積,從而提高合金催化劑活性,即為雷尼效應。Kim等[23]在1993年就提出了雷尼效應,在400~500℃,對Pt-Fe合金進行燒結,然后對電催化劑進行酸處理,使Pt表面未合金化的過渡金屬溶解掉,電催化劑只保留Pt和Pt-Fe合金,Pt的活性表面積增加了兩倍,提高了Pt-Fe合金的催化活性。王彥恩等[24]制備了Pt-Fe合金,采用EDX、XRD和電化學測量技術研究發現有20%的Fe進入了Pt晶格形成Pt-Fe合金,酸處理后其表面積增加30%,氧還原起始還原電位達到0.70V,極限擴散電流密度達到5.15mA/cm2,比純Pt的還原電位0.63V和4.45mA/cm2的 電 流 密 度 高 得 多,提高了Pt的電催化活性。他們同時指出,只有形成合金的Fe能提高氧還原反應的電催化活性,非合金化的Fe對催化性能并無影響,證明了Pt-Fe合金中Fe元素的雷尼效應。Carpenter等[25]采用還原法制備了Pt-Ni合金催化劑,并采用XRD、HAADF-STEM和EDS對晶體組分和形態進行表征,發現不同原子比的Pt-Ni合金的電催化活性是純Pt的3~5倍,且當原子比為1∶1時,Pt-Ni合金的催化活性能達到純Pt的15倍。耐久性測試如圖6所示,進行20000圈CV循環后,PtNi/C合金的比活性從下降到1400μA/,質量比活性由于電化學表面積(ECSA)從26m2/gPt增加到31m2/gPt僅下降40%。雖然催化活性有所下降,但仍能滿足2015年DOE對催化劑性能的要求。ECSA的增加是由于晶粒中非貴金屬的溶解,即與雷尼效應有關。從圖7可以看出,20000圈CV循環后80%的晶粒內部有明顯的凹陷,EDS分析證實這是因為合金中Ni的溶解而造成缺陷。
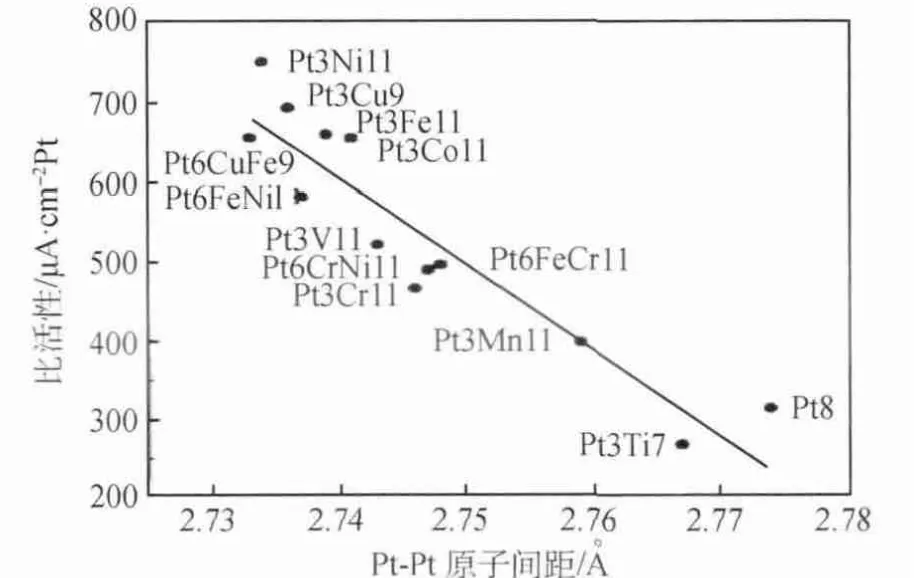
圖5 Pt-Pt的原子間距與合金比活性的關系[21]
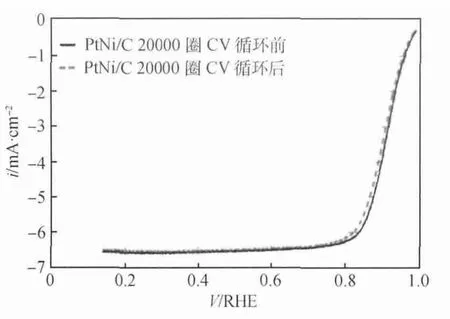
圖6 1600r/min轉速下PtNi/C合金20000圈CV循 環 前 后 的ORR極 化 曲 線[25](電壓范圍0.6~1.0V,速度50mV/s)
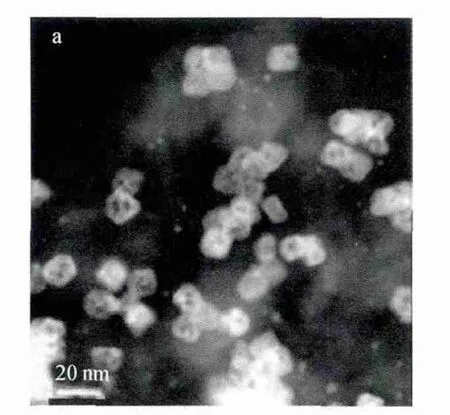
圖7 PtNi/C合金20000圈CV循環后的HAADF圖像[25]
2.4 錨定效應
Pt-M合金催化劑中添加非貴金屬M可使Pt原子更好嵌入或錨定在載體C表面,防止催化劑在C表面集聚或流失,使Pt-M二元或三元合金催化劑具有更高的穩定性,即為合金的錨定效應。魏子棟等[26]早期通過高溫熱處理制備了Pt3-Fe-Co面心立方晶體,Fe和Co位于晶格的面心位置并與載體C之間有較好的相容性,在200mA/cm2的電流密度下工作,發現Pt3-Fe-Co三元合金催化電極陰極還原極化電壓比傳統Pt/C減少20~40mV,比表面積增加了20%,限制C載體表面Pt的遷移,電極性能改善的原因歸因于Fe、Co元素的“錨定效 應”。Mercado等[27]采 用 加 速 耐 久 試 驗 方 法(ADT)對Pt/C和Pt3Ni/C的耐 久性進行測試,觀察到Pt在Pt/C表面遷移,并且Pt顆粒逐漸增大,而Pt3Ni/C合金則不受施加電位的影響,沒有觀察到團狀的金屬顆粒和遷移現象,即過渡金屬M的加入可以防止Pt團聚,將這一現象歸因于Ni對Pt的錨定效應。
3 影響合金催化劑活性的因素
電催化劑的氧還原反應催化活性不僅與添加的3d結構過渡金屬的種類有關,即與催化劑自身的特性有關,還與其表面的堆疊方式有關。
3.1 合金組成
電催化劑氧還原反應催化活性與添加的3d結構過渡金屬的種類有關,研究者對過渡非貴金屬合金PtM/C,如M=Fe、Co、Ni、Cu、V、Ti、Cr和Mn等的性能有很多研究,Mercado等[24]研究發現PtM/C合金(M=Ni、V、Co、Fe)的氧還原活性的大小關系為:PtFe/C>PtCo/C>PtV/C>PtNi/C>Pt/C,且不同原子比的合金的耐久性比純Pt/C要好,不同添加金屬的合金的耐久性能不同,如添加Cr和Ti元素的合金比添加Fe、V、Ni的合金的耐久性能要好[28]。有研究人員解釋為Co和Cr形成合金的合金度更高,Pt-Cr和Pt-Co的原子鍵能更強。在耐久性能測試中,合金催化性能的下降幅度更小,主要歸因于循環電壓下,合金催化劑晶粒長大較小,電催化劑電化學表面積(ECSA)減小較小。如圖8所示[29],經過0.6~1.2V電壓下的CV循環后,Pt/C的晶粒尺寸約增大了5~6倍,而PtCo/C合金的晶粒尺寸并沒有太大的變化,從而從微觀上證明合金催化劑的活性比Pt的活性高。Xiong等[30]研究了不同原子比對Pt-Co合金活性的影響,如圖9所示,為室溫0.65A/cm2的條件下催化劑極化大小與Co/Pt原子比的關系,當Pt∶Co的原子比為1∶7時,合金催化劑具有最小極化,達到最高活性。

圖8 60%Pt/C(上)和40%PtCo/C(下)TEM照片[29]
3.2 合金結構
除了傳統意義的合金催化劑,在納米尺度上對催化劑的結構進行設計和化學裁剪可顯著改善合金催化劑的性能,近年來對多面體結構[31]、納米線結 構[32]和核 殼 結 構[33-34]等 可 控 原 子 的 堆 疊 方 式 有很多研究,以過渡金屬為核,Pt為殼的核殼合金能有效降低Pt的用量,提高催化活性和穩定性。Pt基納米線結構高度有序,有利于電極反應的各相傳質,是良好的電子集流體,可大大提高Pt的催化活性及耐久性[35]。
作者課題組趙天天等[36]采用兩步還原法制備了Co@Pt/C核殼結構催化劑,如圖10(a)所示,實驗發現自制的20%Co@Pt/C(原子比為1∶1)電化學活性比表面積(ECSA)為56m2/g,20%Co@Pt/C(原子比為1∶3)的ECSA為60m2/g,均超過商用20%Pt/C催化劑(ECSA=54m2/g)。其LSV曲線如圖10(b)所示,20%Co@Pt(1∶1)/C、Co@Pt(1∶3)/C和Pt/C的ORR起始電位相 近,半 波 電 位 分 別 為0.831V、0.840V和0.833V,20%Co@Pt(1∶3)/C的電催化活性比商用催化劑有所提升,極大降低Pt用量。崔春華等[37]制備了三種Pt-Ni八面體合金催化劑,實驗發現合金中的富Pt區主要集中在合金晶面的邊緣,而Ni原子則優先排列在{111}晶面上。圖11(a)為在25℃,濃 度 為0.1mol/L的HClO4中,掃描速度為100mV/s下測得CV曲線,圖11(b)為在25℃飽和氧氣,濃度為0.1mol/L的HClO4中,掃描速度為5mV/s測得的LSV曲線,從圖中可以看出,PtxNi1-x八面體合金的ORR活性曲線的半波電位正移73mV,合金活性明顯提高。圖11(c)、(d)中經過3圈和25圈CV循環(電勢范圍為0.06~1.0VRHE,掃描速度250mV/s)穩定后測得的三種合金組分的比活性比Pt/C和Pt納米顆粒催化劑高6~10倍,質量比活性高10~15倍,是目前報道[38-41]的碳載鉑貴金屬合金中質量比活性最高的。
4 結 論
綜上所述,具有3d結構的過渡非貴金屬加入Pt催化劑中,會改變純Pt催化劑的幾何特征和電子層結構,增加Pt表面的粗糙度,對Pt具有錨定作用,并提高催化劑的活性,降低了貴金屬Pt的用量,對燃料電池陰極催化劑活性有顯著提高。研究人員試圖從原子尺度上確定反應的活性中心,理解反應的微觀機理,為工業選擇和設計新的高活性的催化劑提供正確的理論指導。通過計算基元反應的活化能或反應能來尋找ORR中反應速度的控制步驟;量化理論模擬不同金屬及合金的組分和結構,根據催化劑電催化活性與催化劑幾何和電子結構以及制備工藝的定量關系,達到設計新型催化劑的目的。
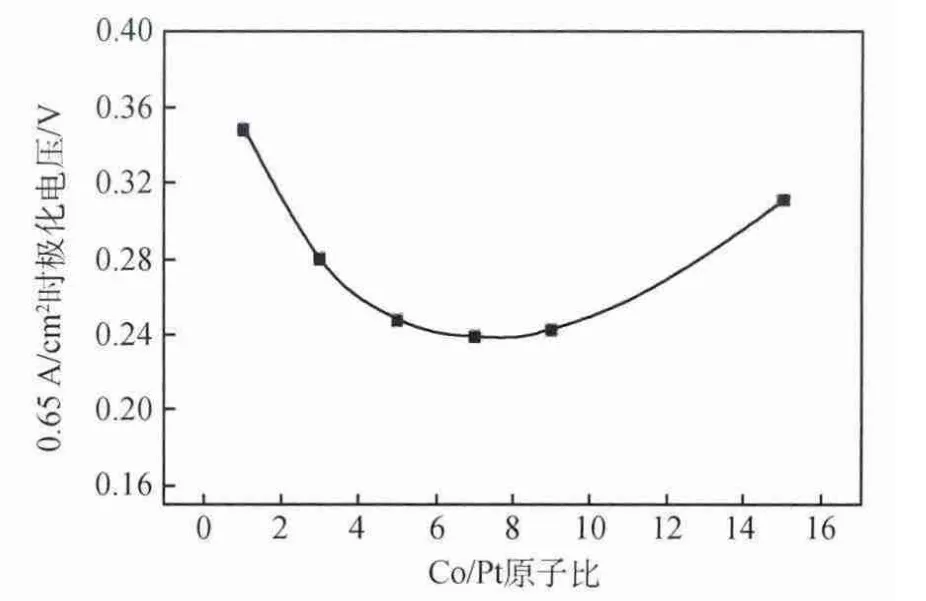
圖9 Co/Pt原子比與催化劑極化大小關系[30]
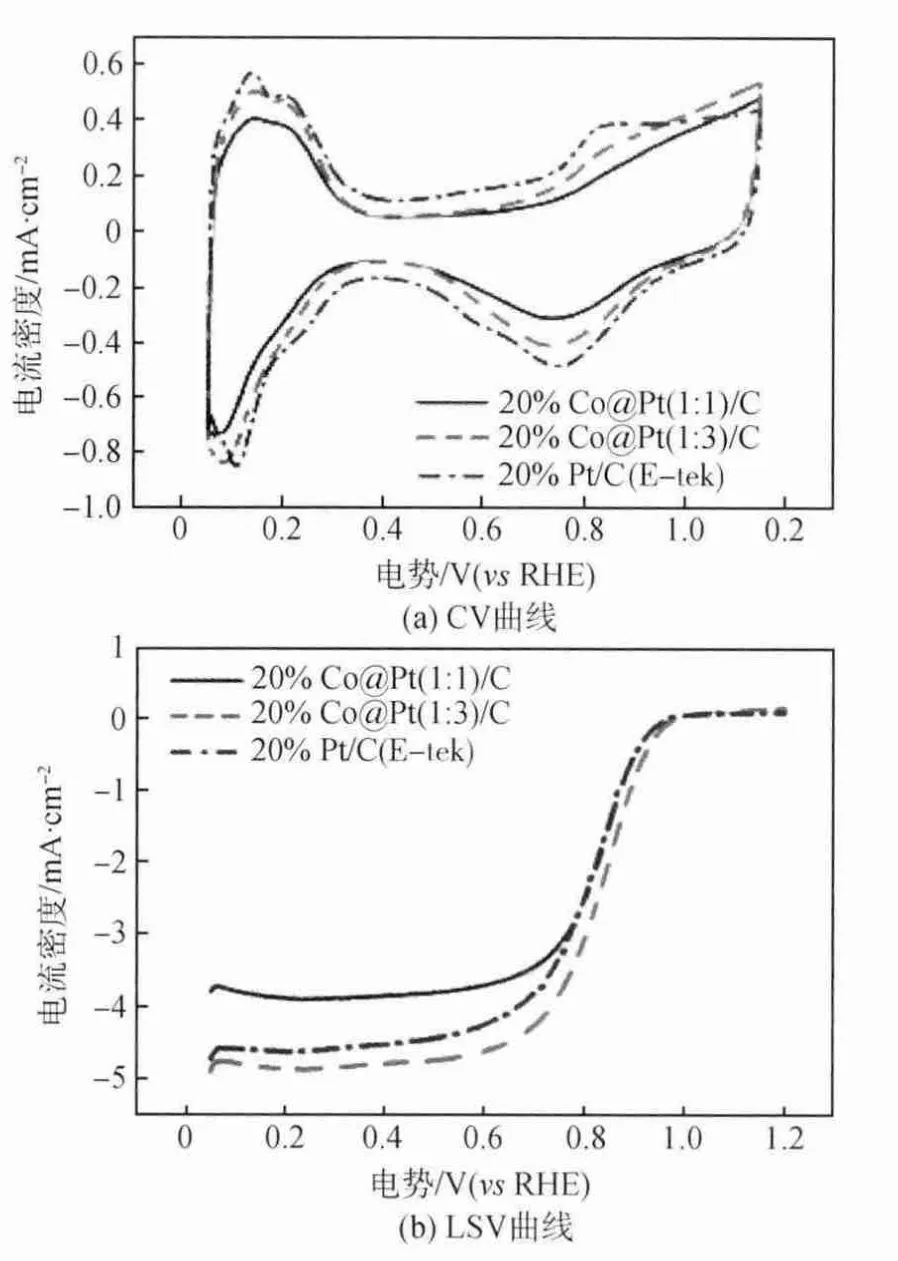
圖10 20%Co@Pt(1∶1)/C、20%Co@Pt(1∶3)/C和20%Pt/C電催化劑的CV曲線和LSV曲線[36]
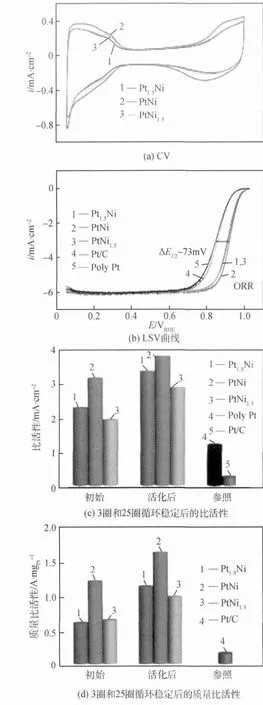
圖11 PtxNi1-x八面體合金的電化學性能
盡管合金化可以提高電催化劑的活性,但是添加元素的溶解會造成合金催化劑耐久性下降,但合金催化劑的催化機理還沒有統一定論,需進一步研究。以往只局限單一因素(電子質子轉移或物種在催化劑表面的脫附吸附)研究ORR機理,沒有系統將兩個因素結合進行模擬研究。對合金的結構特性和制備因素的研究雖然取得了一定的進展,但如何進一步提高合金度,降低Pt的用量,設計和制備出高活性、高穩定性和長壽命的催化劑是催化劑研究的關鍵因素,對合金催化劑催化機理研究具有理論指導意義。
[1] Yuan X Z,Li H,Zhang S S,et al.A review of polymer electrolyte membrane fuel cell durability test protocols[J].J.PowerSources,2011,196:9107-9116.
[2] Greeley J,Stephens I E L,Bondarenko A S,et al.Alloys of platinum and early transition metals as oxygen reduction electrocatalysts[J].NatureChemistry,2009,1:552-556.
[3] 李明芳.鉑電極上氧還原機理與動力學的研究[D].合肥:中國科學技術大學,2011.
[4] 原鮮霞,夏小蕓,曾鑫,等.低溫燃料電池氧電極催化劑[J].化學進展,2010,1:19-31.
[5] 鐘軼良,莫再勇,楊莉君,等.改性石墨烯用作燃料電池陰極催化劑[J].化學進展,2013,5:717-725.
[6] Vliet D,Wang C,Debe M,et al.Platinum-alloy nanostructured thin film catalysts for the oxygen reduction reaction[J].ElectrichimicaActa,2011,56:8695-8699.
[7] Fatih S,Gls N G,Middl E,et al.Improving catalytic efficiency in the methanol oxidation reaction by inserting Ru in face-centered cubic Pt nanoparticles prepared by a new surfactant,tert-octanethiol[J].Energy&Fuels,2008,22(3):1858-1864.
[8] Wang W M,Huang Q H,Liu J Y,et al.One-step synthesis of carbon-supported Pd-Pt alloy electrocatalysts for methanol tolerant oxygen reduction[J].ElectrochemistryCommunication,2008,10(9):1396-139.
[9] Bing Y,Zhang L,Ghosh D,et al.Nanostructured Ptalloy electrocatalysts for PEM fuel cell oxygen reduction reaction[J].Chem.Soc.Rev.,2010,39:2184-2202.
[10] Prakash J,Tryk D A,Yeager E B,et al.Kinetic investigation of oxygen reduction and evolution reaction on lead ruthenate catalysts[J].J.Electrochem.Soc.,1999,146:4145-4151.
[11] Blizanac B B,Ross P N,Markovic N M,et al.Oxygen electroreduction on Ag(111):The pH effect[J].ElectrochimicaActa,2007,52:2264-2271.
[12] Jiang L,Hsu A,Chu D,et al.Oxygen reduction on carbon supported Pt and PtRu catalysts in alkaline solutions[J].JournalofElectroanalyticalChemistry,2009,629:87-93.
[13] N?rskov J K,Rossmeisl J,Logadottir A,et al.Origin of the overpotential for oxygen reduction at a fuel-cell cathode[J].J.Phys.Chem.B,2004,108:17886-17892.
[14] Kalberg G S,Rossmeisl J,Norskov J K,et al.Estimations of electric field effects on the oxygen reduction reaction based on the density functional theory[J].Phys.Chem.Chem.Phys.,2007,9:5158-5161.
[15] Hyman M P,Medlin J W.Mechanistic study of the electrochemical oxygen reduction reaction on Pt(111)using density functional theory[J].J.Phys.Chem.B,2006,110(31):15338-15344.
[16] Wakisaka M,Suzuki H,Mitsui S,et al.Increased oxygen coverage at Pt-Fe alloy cathode for the enhanced oxygen reduction reaction studied by EC-XPS[J].Journal ofPhysicalChemistryC,2008,112:2750-2755.
[17] Lima F H B,Zhang J,Shao M H,et al.Catalytic activity d-band center correlation for the O2reduction reaction on platinum in alkaline solutions[J].JournalofPhysical ChemistryC,2007,111(1):404-410.
[18] Shao M H,Sasaki K,Adzic R R.Pd-Fe Nanoparticles as electrocatalysts for oxygen reduction[J].J.Am.Chem.Soc.,2006,128(11):3526-3527.
[19] Stamenkovic V,Mun B S,Mayrhofer K J J,et al.Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure[J].Angew.Chem.,2006,45(18):2897-2901.
[20] 衣寶廉.燃料電池-原理.技術.應用[M].北京:化學工業出版社,2003:170-179.
[21] Min M,Cho J,Kim H,et al.Particle size and alloying effects of Pt-based alloy catalysts for fuel cell applications[J].ElectrochimicaActa,2000,45:4211-4217.
[22] Kristian N,Yu Y L,Lee J M,et al.Synthesis and characterization of Co core-Pt shell electrocatalyst prepared by spontaneous replacement reaction for oxygen reduction reaction[J].ElectrochimicaActa,2010,56:1000-1007.
[23] Kim K T,Hwang J T,Kim Y G,et al.Surface and catalytic properties of iron‐platinum/carbon electrocatalysts for cathodic oxygen reduction in PAFC[J].J.Electrochem.Soc.,1993:31-36.
[24] 王彥恩,唐亞文,周益明,等.Fe對Pt-Fe/C催化劑電催化氧還原反應活性的影響[J].高等學校化學學報,2007,4:743-746.
[25] Carpenter M K,Moylan T E,Kukreja R S,et al.Solvothermal synthesis of platinum alloy nanoparticles for oxygen reduction electrocatalysis[J].J.Am.Chem.Soc.,2012,134(20):8535-8542.
[26] 魏子棟,郭鶴桐,唐致遠.氧在Pt-Fe-Co/C合金催化劑上的還原[J].催化學報,1995,16:142-144.
[27] Mercado H R,Popov B N.Stability of platinum based alloy cathode catalysts in PEM fuel cells[J].JournalofPower Sources,2006,155:253-263.
[28] Ermete A,Salgadob J R C,Gonzalez E R.The stability of Pt-M(M=first row transition metal)alloy catalysts and its effect on the activity in low temperature fuel cells:A literature review and tests on a Pt-Co catalyst[J].J.PowerSources,2006,160:957-968.
[29] Ball S C,Hudson S L,Leung J H,et al.Mechanisms of activity loss in PtCo alloy systems durability:Cathode catalyst activity and stability[J].ECSTrans.,2007,11:1247-1256.
[30] Xiong L,Kannan A M,Manthiram A.Pt-M(M=Fe,Co,Ni and Cu)electrocatalysts synthesized by an aqueous route for proton exchange membrane fuel cells[J].ElectrochemistryCommunications,2005,4:898-903.
[31] Rabis A,Rodriguez P,Schmidt T J.Electrocatalysis for polymer electrolyte fuel cells:Recent achievements and future challenges[J].ACSCatalysis,2012,2:864-890.
[32] 龔志紅.超細磁性納米線陣列的制備及性能研究[D].南京:南京航空航天大學,2010.
[33] Brushett F R,Duong H T,Wei J,et al.Investigation of Pt,Pt3Co,and Pt3Co/Mo cathodes for the ORR in a microfluidic H2/O2fuel cell[J].Journalofthe ElectrochemicalSociety,2010,157:837-845.
[34] 陳丹,舒婷,廖世軍.核殼結構低鉑催化劑設計制備及核的 組 成 及 結 構 的 影 響[J].化 工 進 展,2013,32(5):1053-1059.
[35] 張敏,李經建,潘牧,等.Pt納米線陣列的氧還原催化性能[J].物理化學學報,2011,27(7):1685-1688.
[36] 趙天天,林瑞,張路,等.Pt含量對Co@Pt/C核殼結構催化劑性能的影響[J].物理化學學報,2013,8:1745-1752.
[37] Cui C H,Gan L,Heggen M,et al.Compositional segregation in shaped Pt alloy nanoparticles and their structural behavior during electrocatalysis[J].Nature Materials,2013,12:765-771.
[38] Carpenter M K,Moylan T E,Kukreja R S,et al.Solvothermal synthesis of platinum alloy nanoparticles for oxygen reduction electrocatalysis[J].J.Am.Chem.Soc.,2012,134:8535-8542.
[39] Snyder J,McCue I,Livi K,et al.Structure/processing/properties relationships in nanoporous nanoparticles as applied to catalysis of the cathodic oxygen reduction reaction[J].J.Am.Chem.Soc.,2012,134:8633-8645.
[40] Wu J,Gross A,Yang H.Shape and compositioncontrolled platinum alloy nanocrystals using carbon monoxide as reducing agent[J].NanoLett.,2011,11:798-802.
[41] Zhang J,Yang H,Fang J,et al.Synthesis and oxygen reduction activity of shape-controlled Pt3Ni nanopolyhedra[J].NanoLett.,2010,10:638-644.
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
