液相—子空間夾角判據聯用分析飲料中的檸檬黃和日落黃
2015-10-09 22:48:54宋光均等
湖北農業科學 2015年17期
關鍵詞:高效液相色譜
宋光均等

摘要:通過液相-光譜聯用,采用向量-子空間夾角判據進行計算,建立了飲料中檸檬黃和日落黃的分析方法。該方法先采用全波長光纖光譜儀建立不同濃度檸檬黃和日落黃的標準光譜數據庫,以及市售碳酸型飲料和功能型飲料的樣品光譜數據,再進行液相-光譜聯用,得到扣除飲料中待測組分后的本底光譜數據;運用向量-子空間夾角判據進行計算飲料中檸檬黃和日落黃的含量。試驗結果表明,在3~30 μg/mL內檸檬黃和日落黃的標準工作曲線線性關系良好(R2檸檬黃=0.997 6,R2日落黃=0.999 8);向量-子空間夾角判據計算結果與高效液相色譜法測定結果比較,相對誤差小于2.5%,樣本加標回收率為89.5%~109.0%,RSD為4.9%~6.1%,該方法能滿足定量分析需求。
關鍵詞:向量-子空間夾角;多波長紫外-可見光譜;高效液相色譜;檸檬黃;日落黃
中圖分類號:O657.3 文獻標識碼:A 文章編號:0439-8114(2015)17-4277-04
DOI:10.14088/j.cnki.issn0439-8114.2015.17.044
檸檬黃和日落黃是碳酸型飲料和功能型飲料中廣泛添加的人工合成色素,由于添加過量色素會對健康造成不良影響,因此建立碳酸型飲料和功能型飲料中檸檬黃和日落黃的分析方法對食品安全檢測具有重要的作用。
目前檸檬黃和日落黃的分析方法有高效液相色譜法[1]、紫外分光光度法[2]、多元線性回歸分光光度法[3]、熒光光譜法[4]、薄層色譜掃描法[5]、示差脈沖極譜法[6]等。檸檬黃含量測定的國家標準(GB4481.1-2010)[7]和日落黃含量測定的國家標準(GB6227.1-2010)[8]都已頒布,各個行業可根據相關國家標準進行檢測,也可在這些技術的基礎上進行改進。隨著儀器技術和信息技術的飛躍發展,分析化學已不再是依賴物質消耗手段來獲取信息,而是逐步發展成為化學信息科學。姚志湘課題組提出了采用“向量-子空間夾角”判據逐步扣減混合光譜中的待測組分光譜,判斷出待測組分恰好消失的“等當點”并計算混合體系中待測組分含量的方法。該方法已經運用于化妝品中對羥基苯甲酸酯、醬油中苯甲酸鈉和山梨酸鉀的測定[9,10]。本研究基于穩健的“向量-子空間夾角”判據,采用液相-光譜聯用技術,建立了飲料中檸檬黃和日落黃的同時分析方法。
1 材料與方法
1.1 儀器與試劑
Maya2000紫外-可見光纖光譜儀(美國海洋光學,掃描波長范圍為190~1 100 nm);LC-20AT高效液相色譜儀(日本島津公司);SZ-93自動雙重純水蒸餾器(上海亞榮生化儀器廠)。檸檬黃(1934-21-0,大連美侖生物技術有限公司);日落黃(2783-94-0,大連美侖生物技術有限公司);乙腈(分析純,安徽時聯特種溶劑股份有限公司);乙酸銨(分析純,西隴化工);飲料(市售)。
1.2 分析方法
1.2.1 標準光譜數據庫的建立 分別精確稱取0.149 5 g檸檬黃、0.150 3 g日落黃對照品于燒杯中,用二次蒸餾水溶解,分別轉移定容至100 mL容量瓶中,得到1 495 μg/mL檸檬黃標準儲備液和1 503 μg/mL日落黃標準儲備液。根據多波長紫外可見光譜儀的響應范圍,用二次蒸餾水依次稀釋標準母液,得到一系列不同濃度的標準溶液,再分別測定一系列不同濃度的檸檬黃標準溶液,日落黃標準溶液的190~1 100 nm紫外可見光譜數據,經最小二乘回歸得到多波長標準光譜數據(v)。
1.2.2 建立市售飲料樣本光譜數據 取一定量的碳酸型飲料,超聲20 min,然后用0.22 μm濾膜過濾。測定各飲料樣品的190~1 100 nm紫外可見光譜(a)。
1.2.3 建立飲料本底光譜數據庫 確定被測組分與本底組分完全分離的高效液相色譜條件,將液相與光譜聯用,采集市售飲料樣本經過色譜柱完全分離后的190~1 100 nm光譜數據,分別扣除功能型飲料和碳酸型飲料樣品中與檸檬黃、日落黃具有相同出峰時間的光譜數據后,得到本底光譜數據(N)。
1.2.4 高效液相色譜條件 色譜柱為Agilent C18柱(5 μm,250 mm×4.6 mm);柱溫為30 ℃;可見紫外光纖光譜儀掃描波長為190~780 nm。流動相梯度∶0.01~9.00 min,乙腈∶0.04 mol/L乙酸銨水溶液=10∶90(V/V);9.01~20.00 min,乙腈∶0.04 mol/L乙酸銨水溶液=40∶60(V/V);20.01~35.00 min,乙腈∶水=10∶90(V/V);35.01~60.00 min,乙腈∶水=95∶5(V/V)。
1.2.5 “向量-子空間夾角判據”數據處理方法 將上述標準光譜數據v,本底光譜數據N、以及樣品光譜數據a導入計算平臺,應用向量-子空間夾角判據算法算出飲料樣本中待測組分的實際含量。具體步驟為:①依據定量精度設定扣減步長Δ;②在函數yi=aix+bi中帶入較大的x1值,得到v1;其中,yi為在i波長下待測組分的吸光度值,ai、bi是常數,x表示檸檬黃或日落黃的濃度,v1表示在濃度為x1時待測組分的多波長吸光度值y1,v1為所有的yi值組成的矩陣;③從所述飲料樣本的光譜數據a中扣除v1/Δ,扣除后的變量為da;把本底光譜數據N和變量da合并后記為對比空間M,計算對比空間M與v1夾角;④從待測飲料樣本光譜數據a中逐步扣除v1后,重復步驟“3”;⑤當所述飲料樣本中的待測組分完全被扣除后比對空間M和待測組分的光譜向量v1空間夾角值會出現最大值θmax,記錄空間夾角最大值θmax出現時對應的扣減步數λ,通過待測組分的濃度x1和扣減步數λ,計算飲料樣本中待測組分的含量Y1,用函數關系定義為:Y1=x1×λ/Δ;⑥比較Y1值與x1值的差值,該差值大于誤差允許范圍,則帶入一個與Y1值相接近的x2進入步驟“2”中重新計算。
1.2.6 加標回收率試驗 在已知濃度的兩份飲料樣品中分別添加一定量的檸檬黃、日落黃標準品,采用“向量-子空間夾角判據”方法測定加標回收率。
1.2.7 精密度試驗 分別準備兩種飲料樣品各5份,采集樣本190~1 100 nm的光譜數據,采用“向量-子空間夾角判據”方法計算樣品中檸檬黃和日落黃含量,計算精密度。
2 結果與分析
2.1 建立標準光譜庫
分別配制系列濃度(3~30 μg/mL)的檸檬黃、日落黃標準溶液,以水為空白對照,分別采集系列溶液的190~1 100 nm光譜,記入標準光譜數據。在檸檬黃標準光譜數據中取450 nm處的各個濃度吸光度進行線性擬合,得到檸檬黃在450 nm處的標準曲線y=0.026 4x+0.026 5,R2=0.999 8;在日落黃標準光譜數據中取480 nm處的各個濃度吸光度進行線性擬合,得到日落黃在480 nm處的標準曲線y=0.041 3x+0.052 4,R2=0.999 8。
2.2 建立市售飲料樣本光譜庫
在全波長下測定各飲料樣品的光譜,構成飲料樣本光譜a。
2.3 建立飲料本底光譜庫
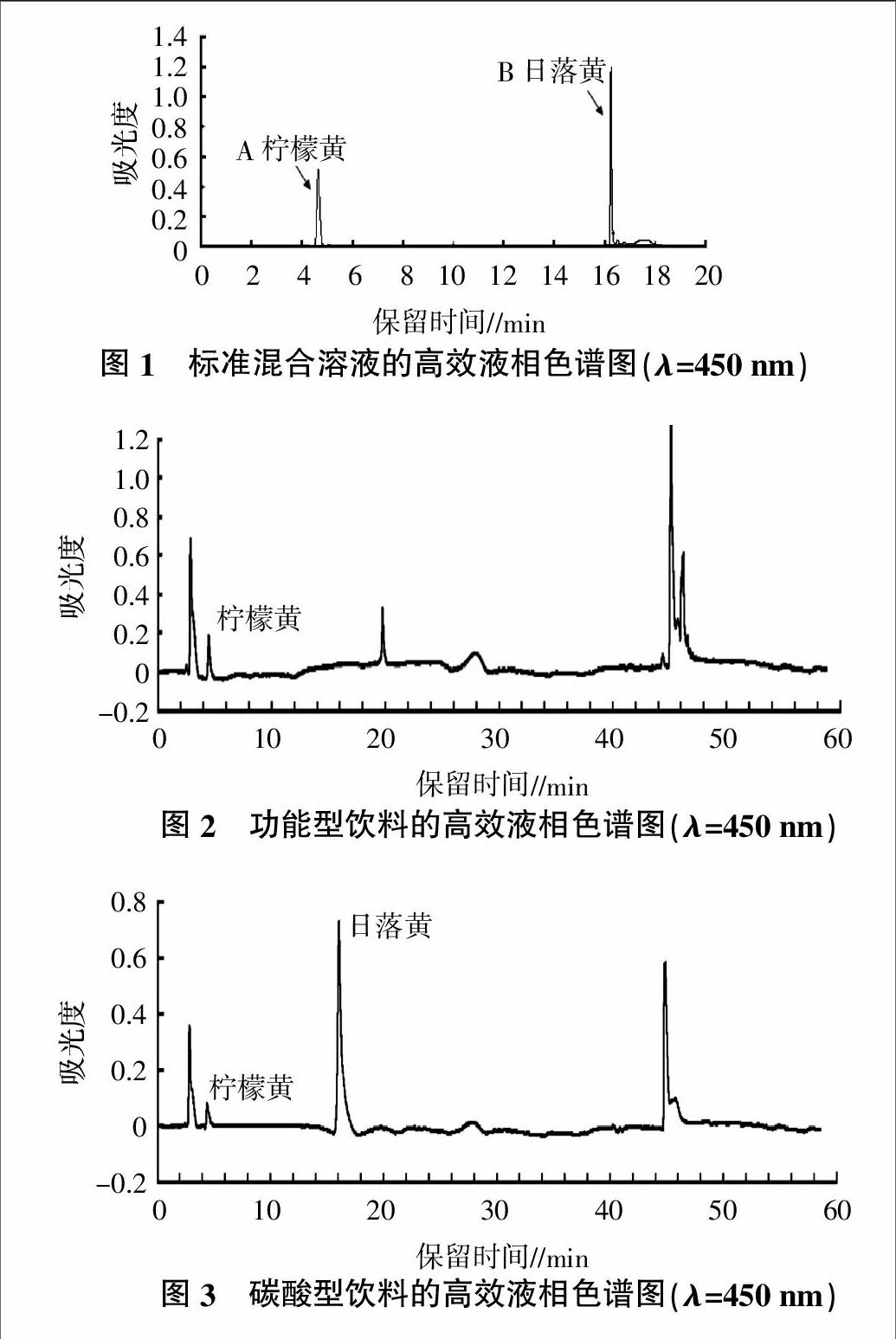
在待測組分與本底組分完全分離的高效液相色譜條件下,得到待測樣本和對照品樣本的高效液相色譜圖。圖1為待測組分對照品在450 nm處的高效液相色譜圖,其中A為檸檬黃、B為日落黃,兩種色素的出峰時間分別為檸檬黃4.678 min、日落黃16.299 min;圖2和圖3為飲料樣本在450 nm處的高效液相色譜圖,經分析對比,樣品色譜圖中A為檸檬黃、B為日落黃,其他的色譜峰為飲料樣本中相對于色素的本底物質。采用液相-光譜聯用采集多波長色譜數據,以檸檬黃的測定結果為例,從功能型飲料的多波長色譜圖中扣除與檸檬黃具有相同保留時間的多波長色譜數據,其余數據為檸檬黃的本底光譜數據;從碳酸型飲料的多波長色譜圖中扣除與檸檬黃具有相同保留時間的多波長色譜數據,其余數據為檸檬黃的本底光譜數據。
功能型飲料和碳酸型飲料的本底光譜數據N1和N2數據量巨大,直接采用N1和N2作為本底數據,計算時間長,運算較困難,影響了方法的時效性。以檸檬黃為例,選用主成分分析[11],在標準化的飲料樣本光譜中添加一定強度的白噪聲蔽不均勻噪聲和非線性因素的干擾,以二階差分值序列的折點判斷獨立變量數目,得到體系主成分數目q。
其中功能型飲料扣除檸檬黃的本底主成分數為22,碳酸飲料扣除檸檬黃后的本底主成分數為21。應用[U,S,V]=svd(N)分別對兩種飲料本底光譜數據庫N1和N2進行奇異值分解降維,經分解后分別得到m階行正交矩陣U、n階列正交矩陣V和奇異值矩陣S,功能飲料本底取U得前22列為降維后的本底數據,碳酸飲料本底取U的前21列,分別重新記作N1、N2。同理,飲料中日落黃含量的分析按檸檬黃的數據處理方法進行本底光譜數據庫的降維。
2.4 市售飲料中檸檬黃、日落黃的測定
將上述的樣品光譜數據、本底光譜數據、檸檬黃和日落黃標準濃度值,以及不同濃度標準光譜數據導入計算平臺,應用向量-子空間夾角判據計算飲料中檸檬黃和日落黃的含量。本研究的方法與高效液相色譜法進行比較,如表1所示。
由表1可知,采用向量-子空間夾角判據計算飲料中檸檬黃和日落黃的含量,與高效液相色譜法測定結果作對比,得到的RSD都小于2.5%,說明向量-子空間夾角判據測定方法準確可靠。
2.5 回收率試驗結果
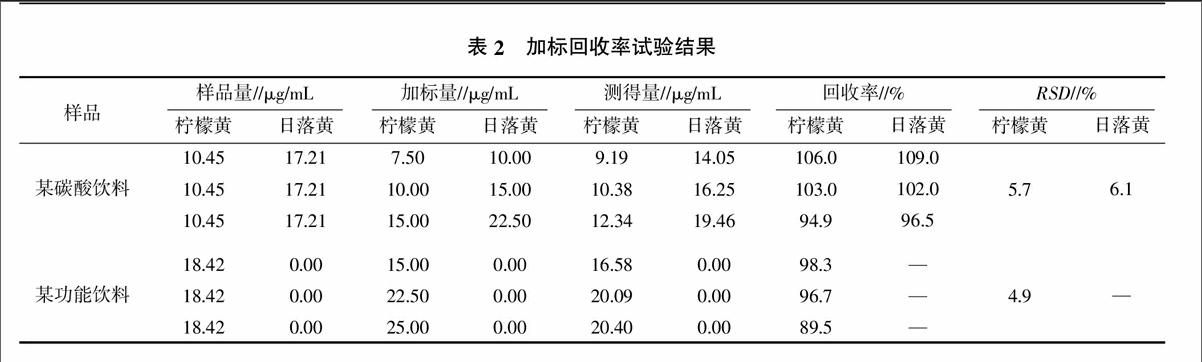
為驗證該方法的準確性,在已知濃度的碳酸飲料中分別添加等體積的檸檬黃、日落黃標準溶液,在另一份已知濃度的功能飲料中添加等體積的檸檬黃標準溶液,然后采用向量-子空間夾角判據計算加標回收率,結果如表2所示。兩種待測組分的樣品加標回收率為89.5%~109.0%,RSD為4.9%~6.1%。表明該方法準確可信,可用于市售飲料的快速檢測。
2.6 精密度試驗結果
采用“向量-子空間夾角判據”計算各樣品中檸檬黃和日落黃含量,試驗結果如表3所示。由表3可知,精密度小于0.35%,說明方法的精密度良好。
3 小結與討論
本研究通過采用液相-紫外可見光譜聯用,分別建立不含被測組分的本底光譜數據、標準光譜數據和待測樣品的一維混合光譜,再通過“向量-子空間夾角”判據進行計算,可解決成分復雜的混合體系中自配本底組分建庫不全的缺點,該方法對于同種樣品的測定,進行一次液相分離后,無需再利用液相-光譜聯用積累本底,僅通過采集樣品的一維光譜,即可實現同類飲料中檸檬黃和日落黃的測定,可降低儀器使用和減小試劑消耗,方法準確性和精密度較好,方法滿足定量分析要求。
參考文獻:
[1] MARLUS C,HERIDA R N S.Validation of a HPLC method for determination of glutamine in food additives using post-column derivatization[J]. American Journal of Analytical Chemistry,2013,(3):113-117.
[2] 劉 冷,李建晴,郭 芬,等.紫外分光光度法同時測定檸檬黃和日落黃[J].光譜實驗室,2007,24(3):423-427.
[3] 陳海春,王宓娜.多元線性回歸分光光度法同時測定檸檬黃和日落黃[J].儀器儀表與分析檢測,2001(4):29-30.
[4] 陳國慶,吳亞敏,王 俊,等.常用合成食品色素熒光光譜研究[J].光譜學與光譜分析,2009,29(9):2518-2522.
[5] 夏立婭,韓媛媛,匡林鶴,等.薄層色譜掃描法同時檢測豆制品中堿性橙、皂黃、檸檬黃和日落黃以及辣椒粉中酸性橙Ⅱ、麗春紅2R和羅丹明B[J].分析試驗室,2010,29(6):15-18.
[6] 李翱楠,郝春香.示差脈沖極譜法測定檸檬黃[J].廣州化工,2011,39(21):112-114.
[7] GB 4481.1-2010,食品安全國家標準食品添加劑檸檬黃[S].
[8] GB 6227.1-2010,食品安全國家標準食品添加劑日落黃[S].
[9] 方 鳳,粟 暉,姚志湘,等.采用向量扣減方法分析化妝品中對羥基苯甲酸酯[J].分析測試學報,2013,32(6):732-736.
[10] 陳 成,粟 暉,姚志湘,等.采用向量-子空間夾角判據測定醬油中的苯甲酸和山梨酸[J].中國食品添加劑,2013(5):223-227.
[11] XIN Q,RUIYAN L,HONGYU Z. Sparse principal component analysis by choice of norm[J]. Journal of Multivariate Analysis,2013,114:127-160.
猜你喜歡
分析化學(2017年1期)2017-02-06 21:32:17
中國醫藥導報(2016年30期)2016-12-28 12:18:02
熱帶農業科學(2016年10期)2016-12-12 01:52:56
分析化學(2016年7期)2016-12-08 00:57:07
上海醫藥(2016年21期)2016-11-21 23:14:07
中國科技博覽(2016年18期)2016-10-19 11:09:28
科學與財富(2016年28期)2016-10-14 04:01:52
中國科技博覽(2016年2期)2016-04-25 14:06:58
上海醫藥(2016年3期)2016-03-23 23:38:20
現代儀器與醫療(2015年4期)2015-07-15 10:13:19
