廣藿香葉片總蛋白雙向電泳體系的建立
2016-05-03 16:21:13何麗平吳友根張軍鋒胡新文
江蘇農業科學 2016年3期
何麗平+吳友根+張軍鋒+胡新文


摘要: 比較2種方法提取廣藿香葉片總蛋白質的得率與聚丙烯酰胺凝膠電泳(SDS-PAGE)蛋白條帶,同時對廣藿香葉片總蛋白質雙向電泳條件進行了優化,利用MALDI-TOF-TOF和Swiss-Prot軟件對部分蛋白點進行質譜鑒定。結果表明:使用TCA/丙酮法提取廣藿香葉片總蛋白,在10萬Vhs聚焦強度下,可獲得高分辨率雙向電泳圖譜,BPP酚抽法獲得的蛋白不適用于雙向電泳分析;經質譜成功鑒定了9個蛋白點,鑒定成功率90%。
關鍵詞: 廣藿香;葉片;雙向電泳;蛋白質提取
中圖分類號: S567.23+9.01 文獻標志碼: A 文章編號:1002-1302(2016)03-0306-04
廣藿香[Pogostemon cablin (Blanco) Benth.]為唇形科刺蕊草屬植物,是我國常用中藥,具有芳香化濁、開胃止嘔、發表解暑等功效[1],是著名國家中藥保護品種藿香正氣丸(膠囊、水)和抗病毒口服液的重要原料,也是其他30多種中成藥的主要原料[2]。但是廣藿香連作障礙現象十分突出,造成產量和品質明顯下降,每茬收獲后必須間種其他農作物后方可再種,嚴重影響了廣藿香規范化生產與種植、藥農種植積極性和區域經濟發展,從而難以保障廣藿香藥材的質量及其標準化、現代化的實施。據報道,廣藿香連作障礙的原因可歸結為根系分泌物質的自毒作用[3]和根際微生物群落多樣性變化[4],但無論何種機制,連作障礙現象都最終在廣藿香植株上表現出來。近年來,隨著現代系統生物學的快速發展,差異蛋白質組學技術逐漸成為研究連作障礙機理的有力工具,其中蛋白質雙向電泳(2-DE)圖譜的建立是進行蛋白質組學分析的前提和基礎,而蛋白樣品的制備則是雙向電泳技術的核心與關鍵。本研究比較不同的廣藿香葉片總蛋白提取與分離方法,旨在為廣藿香葉片差異蛋白質組學分析奠定基礎,進而為揭示廣藿香連作障礙機制提供理論依據。
1 材料與方法
1.1 材料
廣藿香植物材料于2014年4月種植于海南大學園藝園林學院廣藿香種質資源圃。當年11月采集植株葉片,液氮速凍保存,用于總蛋白質提取。
1.2 試劑
三氯乙酸(TCA)、尿素(urea)、硫脲(thiourea)、3-[(3-膽酰胺丙基)-二乙胺]-丙硫酸(CHAPS)、十二烷基磺酸鈉(SDS)、二硫蘇糖醇(DTT)、硫酸銨、考馬斯亮藍G-250,購自美國Sigma公司。24 cm IPG膠條(pH值4~7)和瓊脂糖,購自瑞典GE Healthcare公司。丙烯酰胺(Acr)、甲叉雙丙烯酰胺(Bis)、交聯聚乙烯吡咯烷酮(PVPP),購自美國Bio-Rad Labs公司。其他藥品均為國產分析純,購自北京化工廠。
1.3 方法
1.3.1 蛋白質樣品的制備 分別稱取廣藿香葉片2份,每份1 g,分別放于預冷研缽中,加入適量PVPP,用液氮研磨至粉末,裝入離心管,-80 ℃冰箱中保存待用。廣藿香葉片總蛋白提取流程按照TCA/丙酮法、BPP(四硼酸鈉/交聯聚乙烯吡咯烷酮/苯酚Borax/PVPP/Phenol)酚抽法進行。
1.3.1.1 TCA/丙酮法 參照Wang等提取頑拗植物組織蛋白的方法[5],并略有改動。將1 g材料粉末加入10 mL預冷的含10%TCA、0.07%巰基乙醇的丙酮中,充分混勻后,-20 ℃沉淀過夜,4 ℃、15 000 g離心30 min,棄上清液。加入10 mL 預冷的含0.07 %巰基乙醇的丙酮,混勻后-20 ℃放置1 h,4 ℃、15 000 g離心30 min,棄上清液。以上步驟重復2次。將沉淀用甲醇洗滌2次,丙酮洗滌2次,室溫干燥后溶解于蛋白裂解液(7 mol/L尿素,2 mol/L硫脲,2%CHAPS,13 mmol/L DTT)中,-20 ℃保存備用。
1.3.1.2 BPP酚抽法 采用BPP法[6]提取蛋白質。1 g材料粉末中加入5 mL BPP蛋白提取液[100 mmol/L EDTA,10 mmol/L Tris,50 mmol/L硼砂,50 mmol/L維生素C,1%PVPP(W ∶ V),1%Triton-100(V ∶ V),2% 2-巰基乙醇(V ∶ V)、30%蔗糖(W ∶ V),pH值 8.0]。在室溫下渦旋混勻10 min,再加入等體積的Tris飽和酚(pH值>7.8)。室溫渦旋10 min,4 ℃、15 000 g離心 15 min。轉移上層酚相至另一個新離心管,加入等體積蛋白提取液,室溫渦旋5 min,4 ℃、15 000 g離心15 min。吸取上層清液轉入另一個新離心管,并加入5倍體積冰冷的過飽和硫酸銨甲醇溶液,混勻后在-20 ℃沉淀6 h以上。4 ℃、15 000 g離心15 min,去除上清液,收集沉淀。將沉淀用甲醇洗滌2次,丙酮洗滌2次,室溫干燥后溶解于蛋白裂解液(7 mol/L 尿素,2 mol/L硫脲,2%CHAPS,13 mmol/L DTT)中,-20 ℃ 保存備用。
1.3.2 蛋白定量 蛋白質定量主要參考Bradford的方法[7],在紫外分光光度計上進行蛋白濃度測定,用牛血清蛋白(BSA)作為標準蛋白,繪制標準曲線,測定樣品在波長 595 nm處的吸光度,每個樣品至少重復3次,計算樣品蛋白濃度。
1.3.3 聚丙烯酰胺凝膠(SDS-PAGE)電泳
1.3.3.1 一維凝膠電泳(1-DE) 1-DE主要采用不連續膠SDS-PAGE法[8],用4%濃縮膠和12.5%分離膠進行電泳分離,蛋白梯度上樣分別為10、20、30 μg,電泳參數設置為 5 W/gel 50 min,7 W/gel 2 h。
1.3.3.2 二維凝膠電泳(2-DE) 第1向等電點聚焦在Ettan IPGphorⅡ等電聚焦儀(GE Healthcare)上完成。使用24 cm的pH值4~7線性IPG(GE Healthcare)干膠條,每根膠條上樣量為1 300 mg蛋白,上樣體積455 μL,常溫水化18 h,每根膠條上覆蓋5 mL礦物油防止樣品揮發。采用2種聚焦程序,設定為:250 V,3 h;500 V,2 h;1 kV,1 h;8 kV,3 h;8 kV下進行10萬 Volt hour(Vhs)和11萬 Vhs聚焦,每根膠條限流50 μA。第2向電泳前進行膠條平衡,將聚焦好的膠條放入平衡緩沖液(50 mmol/L Tris-HCl,pH 值8.8,6 mol/L尿素,30%甘油,2%SDS,0.02%溴酚藍)中平衡,先后浸泡在1%DTT的膠條平衡液和含4%碘乙酰胺的膠條平衡液中,各15 min。平衡結束后,轉移膠條垂直放于12.5%聚丙烯酰胺凝膠的上端,用含有適量溴酚藍的1%瓊脂糖封住膠條,保證膠條與凝膠充分接觸,在Ettan Daltsix電泳儀(GE Healthcare)中進行電泳,電泳參數設置為5W/gel 1 h,7 W/gel 3.5 h。
1.3.4 凝膠染色及圖像采集分析 凝膠染色主要采用考馬斯亮藍(G-250)染色液進行染色[9]。脫色干凈的凝膠采用Image ScannerⅢ掃描儀(GE Healthcare)進行掃描,掃描設置為灰階256,投射掃描,分辨率為300 dpi,圖像以TIF格式保存,用ImageMaster 2D Platinum軟件(Amersham Bioscience)進行圖像分析。
1.3.5 蛋白質質譜鑒定 挖取感興趣的蛋白點,使用簡化膠內酶解法進行酶解[10]。酶解得到的肽段混合物質譜鑒定(MALDI-TOF MS)采用AB SCIEX公司的基質輔助激光解析電離飛行時間質譜儀(5800 MALDI-TOFMS/MS)。將獲得的胰蛋白酶解肽段信息通過Mascot引擎搜索NCBI中的綠色植物數據庫,獲得蛋白鑒定信息。
2 結果與分析
2.1 不同提取方法獲得的總蛋白量及SDS-PAGE電泳單向條帶比較
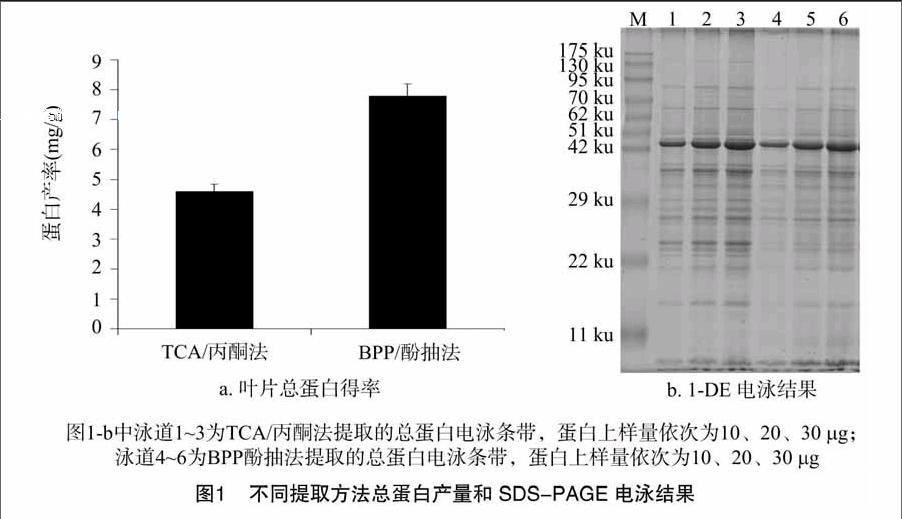
由圖1-a可知,采用TCA/丙酮法可從1 g廣藿香葉片材料中提取4.61 mg蛋白,采用BPP酚抽法可以得到7.79 mg蛋白。利用2種方法提取所得的廣藿香葉片總蛋白SDA-PAGE電泳條帶見圖1-b。結果表明,采用梯度上樣量10、20、30 μg蛋白質樣品進行SDS-PAGE電泳后,TCA/丙酮法與BPP酚抽法提取的蛋白質條帶數量差別不大,但在條帶清晰度上有較大差異。TCA/丙酮法所得蛋白產量低于BPP酚抽法所得蛋白產量,但是其電泳條帶清晰;而BPP酚抽法在30~42 ku 處以及52~75 ku處條帶較為模糊。說明BPP酚抽法所提取的蛋白質純度不夠,含有干擾物質等雜質,可直接影響其蛋白定量和SDS-PAGE電泳過程。
2.2 廣藿香葉片蛋白的2-DE分析
如圖2所示,利用BPP酚抽法得到的樣品蛋白2-DE圖譜中蛋白點數量較少(183個),形狀不圓滑,橫條紋、縱條紋也較多,說明該方法得到的樣品中確實有大量干擾物質存在,與SDS-PAGE電泳蛋白條帶反映的結果一致。所以,盡管通過BPP酚抽法得到的樣品蛋白量較多,但因其質量無法滿足雙向電泳的要求,不能用于廣藿香葉片蛋白質組學分析。TCA/丙酮法得到的2-DE圖譜中蛋白點數量較多(579個),形狀多數圓滑,尤其集中在pH值5~6范圍內。TCA/丙酮法所得圖譜分辨率比BPP酚抽法好,可見TCA/丙酮提取法可減少廣藿香葉片總蛋白質在提取過程中的蛋白質損失,有效去除廣藿香葉片中次生代謝物質等雜質,表現為所獲得電泳圖譜中蛋白質點數量多,蛋白點清晰,無拖尾現象。
對TCA/丙酮法所得總蛋白進行不同聚焦強度比較,由圖3可以看出,聚焦10萬Vhs條件下,中高分子量處蛋白點的聚焦情況有明顯改善,蛋白點的分離效果相對較好,蛋白點數明顯多于聚焦11萬Vhs,這可能與聚焦過度引起部分蛋白點丟失有關。
2.3 質譜鑒定與功能分析
對利用TCA/丙酮法獲得的10個高分辨率蛋白點(圖4)進行質譜鑒定,其中9個蛋白點被成功鑒定(蛋白得分>49),鑒定成功率90%(表1),表明該方法所得蛋白點具有良好的質譜兼容性。蛋白點1為熱激蛋白70,是熱激蛋白家族的一員,在細胞內主要參與新生肽的折疊與成熟、損傷蛋白的降解和蛋白運輸。蛋白點3為預測蛋白——谷胱甘肽 S-轉移酶 L3狀 X1異構亞型,是谷胱甘肽代謝的膜相關蛋白,在植物的初級代謝和二級代謝、脅迫耐受和細胞信號轉導中行使功能,從而影響植物生長發育。蛋白點5為預測蛋白——二氫硫辛酸脫氫酶,它以FAD為輔基,是參與丙酮酸形成乙酰-CoA以及α -酮戊二酸脫氫形成琥珀酰-CoA過程中多酶體系的一種酶。蛋白點10為光合系統Ⅰ反應中心第4單元葉綠體蛋白,其功能是穩定PsaC與光合反應中心之間的相互作用,協助鐵氧還原蛋白與光合系統的對接,與鐵氧還原蛋白——NADP還原酶互作。
3 結論與討論
樣品制備是蛋白質組學研究的重要因素,樣品質量好壞直接決定電泳圖譜進行軟件分析的結果。最優的樣品制備方法應該能有效除去影響蛋白質可溶性和電泳分離結果的各種雜質,防止蛋白變性,保持其活性,減少蛋白質的降解、磷酸化和甲基化修飾,并有高的分辨率和良好的重復性。
然而廣藿香葉片組織中含有大量揮發油、黃酮類、倍半萜類、萜類、蒽醌類等次生代謝物[11-13],這些不利因素將直接導致難以獲得無污染、無修飾、完整、高質量的植物細胞蛋白質,嚴重影響蛋白質的提取效果、等電聚焦及雙向電泳圖譜的分辨率。TCA/丙酮法、BPP酚抽法是2種常用的蛋白質提取方蛋白點7經質譜鑒定,未鑒定成功(蛋白得分小于49分)。匹配肽段數括號內數值代表總匹配肽段數中可信值大于95%的肽段數。
法。TCA/丙酮法可有效沉淀蛋白[14],能夠抑制蛋白酶活性,消除蛋白質水解作用和其他蛋白酶的修飾作用,有效降低蛋白樣品中鹽、糖、脂類、核酸等雜質對試驗的干預[15]。BPP酚抽提法雖然被廣泛應用于植物組織蛋白質提取,但對于廣藿香葉片材料并不適用。在影響等電聚焦的因素中,等電聚焦強度對獲得理想的2-DE圖譜至關重要[16]。不同植物甚至同一植物不同組織的蛋白樣品分離所需的等電聚焦強度不同,同一樣品不同膠條長度所需的等電聚焦強度也有差別。本研究使用的膠條長24 cm,承載的蛋白量多,聚焦充分,蛋白點分離效果好,便于進行蛋白質組學的研究。比較不同聚焦強度下的廣藿香葉片蛋白的2-DE圖譜后,發現在10萬Vhs聚焦強度下,蛋白點最多,可達613個蛋白點,且分離效果好。從2-DE凝膠選取的10個蛋白點中鑒定出9個蛋白點,這些蛋白質參與光合反應、代謝及能量代謝,表明使用TCA/丙酮法提取廣藿香葉片總蛋白,聚焦強度為10萬Vhs時,不僅能得到理想的2-DE圖譜,而且分離的蛋白點具有良好的質譜兼容性。本研究為廣藿香葉片蛋白質組學研究奠定了堅實基礎,也為其他中藥材的葉片總蛋白提取提供了依據。
參考文獻:
[1]國家藥典委員會.中華人民共和國藥典[M]. 北京:化學工業出版社,2009.
[2]吳友根,郭巧生,鄭煥強. 廣藿香本草及引種歷史考證的研究[J]. 中國中藥雜志,2007,32(20):2114-2117,2181.
[3]李玲梅,李 明. 廣藿香根系分泌物的化感自毒作用研究[J]. 湖北農業科學,2011,50(24):5168-5171.
[4]吳友根,郭巧生,鄭煥強. 廣藿香種植土壤和藥材中有機氯農藥及重金屬殘留分析[J]. 中國中藥雜志,2008,33(13):1528-1532.
[5]Wang W,Scali M,Vignani R,et al. Protein extraction for two-dimensional electrophoresis from olive leaf,a plant tissue containing high levels of interfering compounds[J]. Electrophoresis,2003,24(14):2369-2375.
[6]Saravanan R S,Rose J K. A critical evaluation of sample extraction techniques for enhanced proteomic analysis of recalcitrant plant tissues[J]. Proteomics,2004,4(9):2522-2532.
[7]Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding[J]. Analytical Biochemistry,1976,72(1/2):248-254.
[8]Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4[J]. Nature,1970,227(5259):680-685.
[9]Wang X C,Wang D Y,Wang D,et al. Systematic comparison of technical details in CBB methods and development of a sensitive GAP stain for comparative proteomic analysis[J]. Electrophoresis,2012,33(2):296-306.
[10]王旭初,范鵬祥,李銀心. 一種適用于質譜分析的簡化膠內酶解方法[J]. 植物生理與分子生物學學報,2007,33(5):449-455.
[11]Wu Y G,Guo Q S,He J C,et al. Genetic diversity analysis among and within populations of Pogostemon cablin from China with ISSR and SRAP markers[J]. Biochemical Systematics and Ecology,2010,38(1):63-72.
[12]Wu Y G,Li C G,Li X C,et al. Comparison of the essential oil compositions between pogostemon cablin and agatache rugosa used as herbs[J]. Journal of Essential Oil Bearing Plants,2013,16(6):705-713.
[13]Wu L H,Wu Y G,Guo Q S,et al. Comparison of genetic diversity in Pogostemon cablin from China revealed by RAPD,morphological and chemical analyses[J]. Journal of Medicinal Plants Research,2011,5(18):4549-4559.
[14]Damerval C,De Vienne D,Zivy M,et al. Technical improvements in two-dimensional electrophoresis increase the level of genetic variation detected in wheat-seedling proteins[J]. Electrophoresis,1986,7(7):52-54.
[15]Tsugita A,Kamo M. 2-D electrophoresis of plant proteins[J]. Meth Mol Biol,1999,112:95-97.
[16]王鳳茹. 雙向電泳應該注意的幾個關鍵問題[J]. 生物技術通報,2005(6):62-64.
