大花三色堇FPNI—PCR反應體系的優化
2016-07-23 20:02:56李婧曾媛龔勝
江蘇農業科學 2016年5期
李婧++曾媛++龔勝


摘要:融合引物與巢式聚合酶鏈式反應(fusion primer and nested integrated PCR,FPNI-PCR)是一種分離并擴增已知序列旁未知序列的有效方法。以大花三色堇(Viola×wittrockiana Gams.)為材料,為提高FPNI-PCR產物特異性,增加條帶清晰度,降低非特異性條帶的干擾,對FPNI-PCR反應體系中第1輪的模板DNA濃度進行對比,優化了第3輪反應中引物濃度、Taq DNA 聚合酶濃度、緩沖液用量,篩選了第3輪特異性引物的退火溫度,進一步完善了FPNI-PCR反應體系。各因素優化比對試驗表明:FPNI-PCR第1輪反應體系應以稀釋后的DNA為模板,20 μL體系中,用量在4.5~10 ng。第3輪最優反應體系為:20 μL反應體系中,特異性引物濃度0.2 μmol/L,引物SP2濃度 0.75 μmol/L,Taq DNA酶用量1.0 U,dNTP用量2 μL,10×buffer(20 mmol/L,Mg2+ plus)用量2 μL,模板1 μL(第2輪反應稀釋100倍后取1 μL為模板),ddH2O補足。第3輪反應中,退火溫度為64 ℃或66 ℃時條帶最為清晰。
關鍵詞:大花三色堇;FPNI-PCR;體系優化
中圖分類號: Q785文獻標志碼: A[HK]
融合引物與巢式聚合酶鏈式反應(fusion primer and nested integrated PCR,FPNI-PCR)是一種基于熱不對稱交錯PCR (thermal asymmetric interlaced PCR,TAIL-PCR)技術原理的PCR方法[1],主要是利用目標序列旁的已知序列設計3 個嵌套特異性引物,與給出的9個特殊設計的隨機簡并引物(arbitrary degenerate prime,AD)相結合,進行1次巢式反應,并在簡并引物的基礎上設計2個嵌套的特殊引物替換第1輪PCR所用隨機引物用于第2、3輪的擴增。FPNI-PCR中融合引物(fusion primer),即特殊設計的隨機簡并引物,由3′端的隨機寡核苷酸(arbitrary degenerate oligonucleotides)和5′端可設計特異引物的一段序列組成,該序列用以替換初次PCR所用簡并引物。經過熱不對稱循環后的產物含有特異引物結合位點,能有效降低產物稀釋后非特異性擴增的影響[2]。FPNI-PCR具有高效靈敏、產物特異性高、重復性好、能夠在較短的時間內獲得目標片段等優點,是擴增基因側翼序列特別是啟動子的有效方法[3]。
FPNI-PCR與TAIL-PCR技術類似,其反應產物的穩定性、可重復性明顯受到反應體系模板、引物、Taq DNA 聚合酶、Buffer以及3輪退火溫度等因素的影響[4]。在進行 FPNI-PCR 反應獲取未知序列時,有必要根據上述因素,對不同因素進行優化組合,以獲得最優的反應結果。
大花三色堇(Viola×wittrockiana Gams.)是重要的冬春季花卉,有著十分豐富的花色和花斑類型,是研究植物花斑形成的理想植物[5]。筆者所在實驗室先前的研究表明,二氫黃酮醇還原酶(dihydroflavonol reductase,DFR)和花青素合成酶 (anthocyanidin synthase,ANS)基因是三色堇花斑由無色轉向著色的關鍵性基因[6]。關于DFR和ANS的相關研究發現,其啟動子中含有的眾多調控元件,與花色素調控密切相關[7-9]。因而這2個基因的啟動子結構特點,可能是花斑位置形成的一個關鍵性因素,其啟動子的克隆,對于進一步研究三色堇色斑的形成具有十分重要的意義。FPNI-PCR技術是克隆啟動子序列的有效方法,而目前關于三色堇FPNI-PCR技術研究尚無相關報道,本研究以三色堇為材料,對其FPNI-PCR反應體系進行優化,為三色堇VwDFR和VwANS基因啟動子的克隆提供技術基礎。
1材料與方法
1.1材料與試劑
大花三色堇品種“賓哥”,花色為黃底黑斑,栽培于海南大學園藝園林學院基地。
1.2方法
1.2.1模板DNA的提取
提取三色堇幼葉DNA,步驟參考尚嘯等的改良CTAB法[10],僅核酸分離時改用加1/10體積的5 mol/L NaAc。提取的DNA用0.8%的瓊脂糖凝膠電泳檢測DNA質量,電泳結果通過凝膠成像儀(Dolophin- Doc,美國)拍照,利用紫外分光光度計測濃度。小管分裝-20 ℃保存。
1.2.2試驗中用到的引物包括FPNI-PCR反應體系中的第1輪、第2輪、第3輪的簡并引物、特殊引物(表1)[1]以及根據VwANS基因設計的特異引物(表2)。
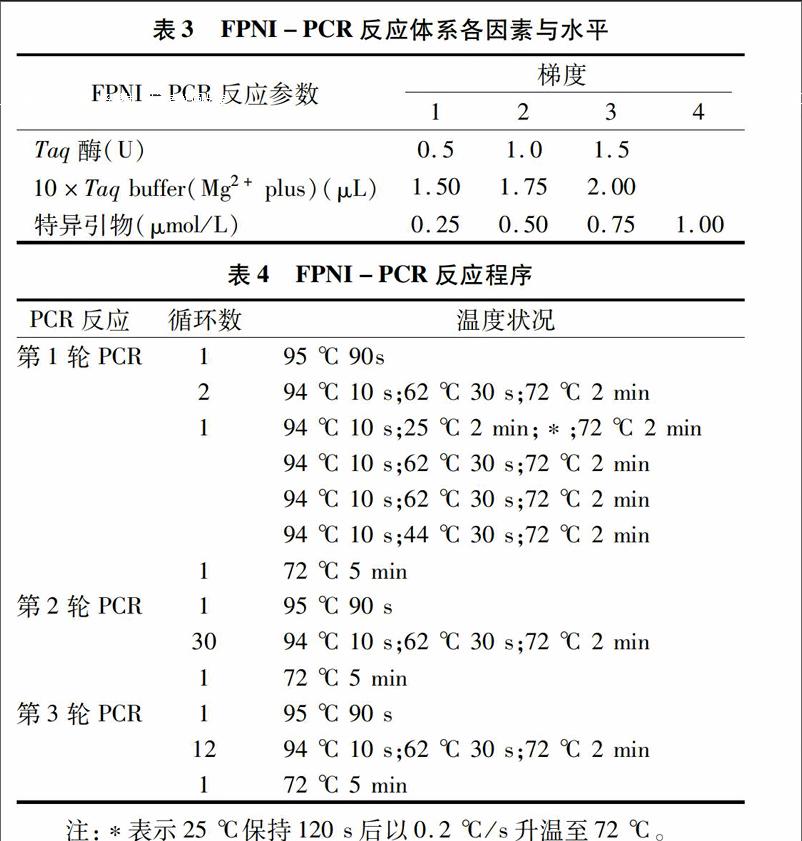
1.2.3FPNI-PCR體系的優化(1)模板DNA濃度篩選。將提取的DNA稀釋100、200倍,然后將未稀釋的DNA和稀釋不同倍數的DNA作為第1輪PCR反應的模板,反應采用表1中的FP5、FP6、FP7、FP9這4條簡并引物,反應程序見表3,反應體系為20 μL,具體組分見參考文獻。
(2)FPNI-PCR反應退火溫度的梯度篩選。根據設計的特異性引物GSP3的TM值,對第3輪退火溫度進行梯度篩選,篩選溫度分別是62、64、66 ℃。
(3)引物、10×buffer(20 mmol/L,Mg2+ plus)、Taq DNA酶用量的對比。 PCR反應采用20 μL反應體系,分別對影響第3輪反應體系中的主要參數:引物、10×buffer(Mg2+ plus)、Taq DNA酶,設置不同的濃度梯度,具體參數值見表3。此外,每20 μL反應體系中均含有dNTP(2.5 mmol/L)2 μL、特異性引物0.2 μmol/L,模板為第2輪反應產物稀釋100倍取1 μL,加ddH2O補足至20 μL。逐一優化每個參數值,優化時,其他參數值均采用表3所列反應參數的第一水平值,引物采用參考文獻中的FP5、FP6、FP7、FP9這4條簡并引物 (表1),反應程序見表4。
2結果與分析
2.1DNA的提取結果
以三色堇嫩葉為材料提取DNA,經紫外分光光度計檢測,濃度為(0.901±0.019)μg/L,D260 nm/D280 nm介于1.8~2.0之間,說明樣品DNA中雜質較少。0.8%瓊脂糖凝膠電泳結果如圖1所示,點樣孔無滯留,條帶清晰明亮,無明顯拖尾現象,說明RNA等雜質去除較干凈。結果表明,采用改良CTAB法可以有效地提取三色堇幼葉的DNA[10],對于后續PCR反應有利。
2.2DNA的濃度對FPNI-PCR的影響
模板DNA的用量對FPNI-PCR反應有著非常直觀的影響,過多或者過少都會造成產物的不穩定或者無法產生條帶。普通方法提取的DNA通常含有雜質,而雜質對PCR產物的影響是非常顯著的。為了獲得清晰條帶,降低非特異性擴增,本試驗以3個濃度的DNA為模板,利用FP5、FP7、FP9這3種簡并引物分別擴增,其他條件相同,結果如圖2所示。
以未稀釋DNA為模板,3輪擴增后均無清晰條帶(圖2)。將DNA模板分別稀釋100、200倍,隨著DNA稀釋倍數的增加,簡并引物FP5擴增出的條帶數量明顯增加,且條帶清晰度明顯提升;簡并引物FP9擴增的結果,100倍稀釋比200倍稀釋擴增條帶數量更多、條帶更加清晰(圖2)。同時,7號引物在多個模板濃度下皆無明顯條帶,說明利用FPNI-PCR進行擴增時,模板的稀釋倍數應配合不同的簡并引物進行篩選,以期降低雜質等對反應的影響,才能獲得更為清晰的條帶。
2.3退火溫度對擴增產物的影響
通過對5、7、9號引物擴增反應的第3輪擴增的退火溫度進行篩選,篩選溫度為62、64、66、68 ℃,結果見圖3。總體來看,64、66 ℃反應效果更好,但是不同引物反應的最佳退火溫度不同,如FP5引物64 ℃下擴增效果較好,FP9號中超過700 bp 的條帶,在66 ℃下更清晰。
2.4Taq酶的用量對FPNI-PCR的影響
Taq酶在PCR反應體系中用量少,卻極為重要,Taq酶的類型、用量,甚至是生產商,都會對PCR產物造成明顯的影響。在本研究中,分別對以FP6和FP7簡并引物擴增獲得的PCR產物進行Taq酶的優化,其他條件全部相同,結果如圖4所示。試驗發現,Taq酶的用量對反應條帶的數量和清晰度都有一定影響,隨著Taq酶用量的增加,FP6和FP7擴增的PCR產物的量均有所提高,但FP6更為明顯,能獲得較為清晰的條帶,而FP7無明顯的清晰條帶。以簡并引物FP6擴增,Taq酶用量1.5 U時比1.0 U時條帶亮度只略有提升,且非特異性擴增也更為明顯,表明Taq酶用量1.0 U更為合適。
2.510×Taq buffer(Mg2+ plus)用量對FPNI-PCR的影響
分別以簡并引物FP6和FP7進行擴增,通過改變10×Taq buffer(Mg2+ plus)用量,其他條件均相同,結果見圖5。結果顯示,以簡并引物FP6擴增,3種緩沖液用量均獲得條帶,但用量為2 μL時,目的條帶更為清晰;以簡并引物FP7擴增,緩沖液為1.5 μL時無條帶,由1.75 μL增至2 μL時,條帶數量增加,清晰度略有增加。
2.6引物濃度對FPNI-PCR的影響
以FP6和FP7這2種簡并引物擴增,對第3輪引物SP2濃度進行篩選。結果(圖6)顯示,引物濃度為0.25 μmol/L時,均無條帶,隨著引物濃度的上升,條帶數量、亮度明顯增加,非特異性擴增同樣增加,條帶出現模糊現象。由圖6可知,以FP6和FP7為簡并引物擴增,第3輪引物SP2濃度為0.75~1.0 μmol/L時,均有較好結果。但從節約成本、提高條帶清晰度、減少非特異性擴增角度考慮,0.75 μmol/L時更佳。
3結論與討論
3.1結論
從試驗結果來看,FPNI-PCR適合以低濃度DNA為模板,第1輪反應以稀釋后的DNA為模板,20 μL體系中,用量在4.5~10 ng。對三色堇FPNI-PCR第3輪體系進行優化,最優反應體系是:20 μL反應體系中,特異性引物濃度 0.2 μmol/L,引物SP2濃度0.75 μmol/L,Taq DNA酶用量10 U,dNTP用量2 μL,10×buffer(20 mmol/L,Mg2+ plus)用量2 μL,模板為第2輪反應稀釋100倍后取1 μL,ddH2O補足。第3輪反應經溫度篩選,退火溫度64 ℃或66 ℃時條帶最佳。
3.2討論
FPNI-PCR是基于熱不對稱PCR技術原理的PCR方法,共包括3輪PCR反應。其中,第1輪反應中,含3次高嚴謹反應,目的是使高退火溫度的特異性引物SP1與模板序列退火延伸;1次低嚴謹反應,目的是使簡并引物結合到較多的目的序列上;5次熱不對稱的高低特異性循環交替反應,使目的片段得以指數性擴增。第2、3輪為普通PCR反應,通過特異性嵌套引物進一步擴增目的條帶。它與普通TAIL-PCR的主要區別在于給出了經過優化后的一系列簡并引物,并在簡并引物的基礎上設計了嵌套的特殊引物FSP1、FSP2用于第2、3輪的擴增[2]。特異性嵌套引物的存在,使非目的條帶得不到大量擴增,而非目標序列中的發卡結構也能抑制其擴增,從而提高了目的片段擴增的準確性[11]。
由于FPNI-PCR反應步驟較多,因此影響擴增結果的因素也較多。首先,第1輪的高低嚴謹熱不對稱反應是目標片段擴增的最初環節,而本試驗證明,模板濃度是影響第1輪反應結果的關鍵因素。在研究中發現,低濃度的DNA模板更容易獲得擴增結果,且模板DNA的稀釋倍數對不同簡并引物擴增的影響不同。在其他PCR反應的研究中同樣發現,植物DNA模板的濃度對于目的片段的擴增具有很大的影響[12]。在TAIL-PCR中,通常需要對模板進行稀釋,才能獲得較為清晰的條帶[4,13]。原因可能是DNA中所含雜質的影響,未稀釋的高濃度DNA中所含雜質更多,影響條帶擴增,另外,不同簡并引物對模板的濃度要求也可能不同,所適應的模板濃度存在差異。
在FPNI-PCR反應中,第1輪擴增產物因條帶雜、濃度低,往往無法通過凝膠成像顯示,需要通過第2輪對目標條帶的進一步擴增獲得初步結果。通過第2輪產物凝膠成像,以有條帶的第2輪稀釋產物為模板,通過第3輪反應進一步擴增,以期獲得較第2輪更為清晰的條帶。因此,第3輪反應體系的優化對最終結果的獲得具有重要作用。
本研究中,針對第3輪PCR反應中部分因素進行了優化,結果發現,與buffer(Mg2+ plus)、Taq DNA酶用量相比,引物的濃度影響最為顯著,隨著引物濃度的上升,條帶數量、亮度明顯增加。需要注意的是,較高的引物濃度雖有利于擴增,但會造成條帶模糊、非特異性條帶增加,這與其他PCR反應類似[14],不利于與第2輪條帶比對推測目的條帶。雖然 FPNI-PCR中融合引物特殊且相對較短,有利于與DNA模板結合擴增目的條帶,但經過3輪反應雖然在一定程度上降低了非特異條帶的干擾,但雜條帶的影響仍是影響試驗結果的重要原因[15]。因此,在第3輪反應中選出最適當的引物濃度,具有十分重要的作用。含Mg2+ 的buffer緩沖液主要對引物和模板的退火、Taq DNA酶的穩定起作用,當緩沖液低于一定限度不能產生條帶,但用量過多也會使dNTP過量結合,非特異性條帶增加,不利于反應[16-17]。與緩沖液相對應,Taq DNA酶用量的多少,同樣對PCR反應存在直接影響,不同的Taq酶需要配合相應的緩沖液使用,過少無法產生條帶,過多也會造成非特異性擴增[18-19]。在本試驗中,較高的buffer濃度和Taq DNA酶均有利于擴增。
PCR反應中除各因素外,退火溫度對目的條帶的獲取同樣具有重要作用。相關研究發現,對熱不對稱PCR第3輪反應退火溫度的優化,能有效地促進目的片段的富集[20]。本試驗第3輪PCR反應中,特異性引物的Tm值為65 ℃,通過溫度篩選得出,退火溫度為64 ℃或66 ℃時,條帶更為清晰。
綜上所述,本研究發現,在FPNI-PCR中通過優化反應體系中各因素的濃度或用量,篩選3輪PCR的退火溫度,均能有效地增加目的條帶的清晰度,減少非特異性擴增,為后續研究打下良好基礎,為其他物種基因啟動子的研究提供一定的借鑒作用。
[HS2*2][HT8.5H]參考文獻:[HT8.SS]
[ZK(#]Wang Z,Ye S F,Li J J,et al. Fusion primer and nested integrated PCR (FPNI-PCR):a new high-efficiency strategy for rapid chromosome walking or flanking sequence cloning[J]. BMC Biotechnology,2011,11(1):16697-16702.
[2]李昆鵬,朱化彬,郝海生,等. 基于PCR方法的基因序列全長獲取策略[J]. 中國生物工程雜志,2012,32(11):115-123.
[3]方子義. 蠟梅查爾酮合成酶基因(CHS)啟動子及相關轉錄因子的克隆與功能分析[D]. 武漢:華中農業大學,2014.
[4]鄭岑,張立平,唐忠輝,等. TAIL-PCR技術及其在植物基因中的克隆[J]. 基因組學與應用生物學,2009,28(3):544-548.
[5]張其生,包滿珠,盧興霞,等. 大花三色堇育種研究進展[J]. 植物學報,2010,45(1):128-133.
[6]李琴. 三色堇花色素合成結構基因的克隆與表達差異分析[D]. 海口:海南大學,2013.
[7]唐杏姣,韓科廳,胡可,等. 菊花CmDFR與CmANS基因啟動子序列克隆與瞬時表達分析[J]. 生物技術通報,2012,28(5):81-88.
[8]Dong W,You Y X,Niu L L,et al. Isolation and analysis of the promoter of an anthocyanin synthase gene from purple-fleshed sweet potato tubers[J]. Acta Physiologiae Plantarum,2014,36(10):2637-2649.
[9]Gollop R,Even S,Colova-Tsolova V,et al. Expression of the grape dihydroflavonol reductase gene and analysis of its promoter region[J]. Journal of Experimental Botany,2002,53(373):1397-1409.
[10][ZK(#]尚嘯,王健,龔勝,等. 三色堇葉片DNA不同提取方法比較[J]. 湖北農業科學,2014,53(20):4999-5002.
[11]劉慧,劉貫山,劉峰,等. 煙草T-DNA插入位點側翼序列擴增方法的篩選與優化[J]. 中國煙草科學,2014,35(1):96-101,107.
[12]李宗菊,熊麗,桂敏,等. 非洲菊基因組DNA提取及[ISSR-PCR擴增模板濃度優化[J]. 云南植物研究,2004,26(4):439-444.
[13]羅靜瑤,陳云云,謝俊,等. TAIL-PCR的技術改良及對香蕉[WTBX][STBX]NBS-RGC5[WTBZ][STBZ]基因側翼序列的克隆[J]. 廣東農業科學,2013,40(10):143-145,170.
[14]劉陽,楊淑霞,李敏惠,等. 引物濃度與退火溫度不當導致巢式PCR非特異性擴增[J]. 成都醫學院學報,2008,3(2):111-114.
[15]胡丹. 月季“月月紅”酵母單雜交cDNA文庫及RrMYB7誘餌載體的構建[D]. 武漢:華中農業大學,2014.
[16]Liu X D,Zheng D,Zhou Y N,et al. Restriction endonucleases digesting DNA in PCR buffer[J]. Journal of Forestry Research,2005,16(1):58-60,85-86.
[17]王云,秦偉,王永豐. 新疆野蘋果S基因特異性PCR體系優化[J]. 新疆農業科學,2013,50(11):2023-2030.
[18]李謀強,師桂英,葉樹輝,等. ‘蘭州百合ISSR-PCR體系的優化[J]. 甘肅農業大學學報,2014,49(6):76-81.
[19]周雨晴,杜沛霖,傅鵬,等. 青天葵ISSR-PCR體系優化及引物篩選[J]. 中國實驗方劑學雜志,2014,20(21):95-99.
[20]陳軍營,沈向磊,辛玉茹,等. 改良TAIL-PCR技術在小麥PM H+-ATPase基因啟動子克隆中的應用[J]. 河南農業大學學報,2008,42(1):1-5.
