土壤中10種多溴聯苯醚的加速溶劑—固相萃取凈化方法優化研究
2016-10-21 16:13:32相雷雷宋洋卞永榮生弘杰柳廣霞蔣新李國華王芳
分析化學 2016年5期
關鍵詞:氣相色譜
相雷雷 宋洋 卞永榮 生弘杰 柳廣霞 蔣新 李國華 王芳

摘要:建立了土壤中10種多溴聯苯醚(PBDEs)的加速溶劑萃取固相萃取凈化氣相色譜分析測定方法。采用加速溶劑萃取(ASE)技術對土壤中10種PBDEs進行提取, 并對4種萃取體系(正己烷、正己烷丙酮(4∶1, V/V)、正己烷丙酮(1∶1, V/V)、正己烷二氯甲烷(1∶1, V/V))進行優化;采用固相萃取(SPE)技術對樣品進行凈化, 制備了10種不同填料的SPE柱, 通過洗脫實驗和加標回收率實驗對各SPE柱的凈化性能進行對比篩選。最終優化條件為正己烷丙酮(4∶1, V/V)體系提取, 酸性硅膠柱凈化。在優化條件下, 10種PBDEs 的回收率為74.4%~125.2%, 相對標準偏差為4.4%~14.4%, 方法檢出限為0.04~0.22 ng/mL。本方法簡單、快速、凈化效果較好、重現性和回收率良好, 可用于土壤樣品中PBDEs的分析。
關鍵詞 :土壤;多溴聯苯醚;加速溶劑萃取;固相萃取凈化;氣相色譜
1 引 言
多溴聯苯醚(Polybrominated diphenyl ethers, PBDEs)作為一類溴代阻燃劑(BFRs), 廣泛應用于電子、紡織、建材和家具等工業產品。PBDEs屬于持久性有機污染物(POPs), 具有疏水性, 易于在顆粒物和沉積物中吸附[1]。PBDEs在環境中難降解, 滯留時間長。大氣、水體、土壤中的PBDEs可通過“蚱蜢跳效應”廣域遷移, 導致全球污染[2]。毒理學研究表明, PBDEs在動物和人體中會長期累積, 并通過食物鏈和生物放大作用向人體轉移, 影響甲狀腺[3~7]、內分泌及神經[7~9]等系統的正常功能, 同時可能存在潛在的致癌性[10]。
目前,對土壤樣品中PBDEs的提取方法有索氏萃取[11~13]、超聲波輔助萃取[14]、微波輔助萃取[13]、加速溶劑萃取等[13~19]。索氏萃取法費時, 且有機溶劑消耗量大;超聲波和微波萃取法可節省提取時間和溶劑, 但提取不完全[11,14]。加速溶劑萃取技術(Accelerated solvent extraction, ASE)具有操作簡便、萃取效率高、速度快、有機溶劑用量少等特點, 是一種省時、安全、自動化的萃取技術, 廣泛應用于土壤中農藥殘留[15]、多氯聯苯[13,16]、多環芳烴以及多溴聯苯[12,14,16,19,20]等污染物的分析檢測。
ASE土壤提取液成分復雜, 雜質較多, 須進行凈化處理。本研究將固相萃取技術(Solid phase extraction, SPE)應用于土壤樣品的凈化。SPE樣品前處理技術具有高效、快速、方便和高選擇性等優點, 被廣泛應用于環境樣品分析的前處理過程中[21~27]。目前, 土壤介質中PBDEs的凈化存在過程復雜、有機溶劑用量大、靈敏度低、重現性差等問題[14,28,29]。選擇合適的填料是提高除雜效率、獲得良好的回收率及重現性的關鍵, 因此, 本研究重點優化了ASE提取和SPE純化條件, 并結合氣相色譜電子捕獲法(Gas chromatography with electron capture detector, GCECD), 建立一種高效、快捷、高靈敏度且具有低檢出限的土壤中PBDEs分析方法。
2 實驗部分
2.1 實驗試劑
正己烷、二氯甲烷(DCM)、丙酮(色譜純,美國Merck公司);硅藻土(100~200目, 德國Fluka公司);弗羅里硅土(60~100目, 美國TEDIA公司);無水Na2SO4、Al2O3(100~200目)、硅膠(100~200目)、石英砂、H2SO4(分析純)購于國藥集團化學試劑公司;實驗用水為去離子水。
PBDEs標準樣品:BDE15, BDE28, BDE47, BDE66, BDE77, BDE99, BDE100, BDE153, BDE154, BDE183, 濃度為1000 ng/mL, 購自美國AccuStandard公司。
2.2 供試土壤
2.3 PBDEs標準曲線的繪制及檢出限的確定
采用Agilent 7890A GCECD(美國Agilent公司), 對PBDEs進行定性與定量分析。色譜條件:DB5色譜柱(30 m × 0.32 mm × 0.25 μm), 進樣口溫度為265℃, 載氣為高純氮氣, 流量為2 mL/min, 檢測器溫度為298℃, 進樣量為1 μL, 不分流進樣。升溫程序:初始溫度140℃, 保持2 min, 5℃/min升至180℃, 保持5 min;5℃/min 升至265℃, 保持5 min;15℃/min升至315℃, 保持10 min。
配制濃度為10, 25, 50, 100, 250和500 ng/mL的PBDEs混標溶液, GC測定。以進樣濃度為橫坐標, 峰面積為縱坐標, 繪制標準曲線。同時, 以信噪比S/N=3時對應的濃度作為儀器的方法檢出限。
2.4 固相萃取柱制備及洗脫實驗
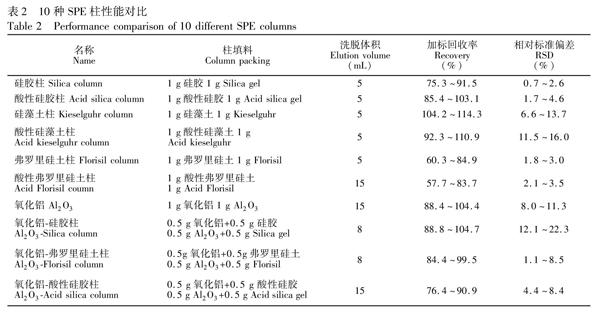
選取實驗室常用的硅膠、弗羅里硅土、硅藻土、氧化鋁4種填料, 并通過濃H2SO4改性制備了酸性硅膠、酸性弗羅里硅土、酸性硅藻土3種改性填料。SPE柱裝填順序(自下而上)為墊片、0.5 g無水Na2SO4、填料層、1 g無水Na2SO4及墊片。根據不同填料的單一及復配組合, 共制備了10種SPE柱(表2)。
洗脫實驗:用5 mL正己烷活化SPE柱后向柱中加入40 μL 1000 ng/mL PBDEs混合標準溶液, 移取1 mL正己烷進行洗脫, 以進樣瓶接收洗脫液, 待洗脫液完全過柱后更換進樣瓶。重復以上操作, 直至洗脫液總體積達18 mL。洗脫液氮吹定容至0.5 mL, GC測定。以洗脫體積為橫坐標, 總回收率(累積求和)為縱坐標, 繪制洗脫曲線。
2.5 PBDEs土壤提取液凈化實驗
土壤中PBDEs提取:準確稱取1.00 g供試空白土壤于燒杯中, 加入40 μL 1000 ng/mL PBDEs混標溶液, 待溶劑揮發后加入2 g硅藻土, 攪拌均勻后裝入不銹鋼萃取池, 采用ASE200型加速溶劑萃取儀(美國DIONEX公司)進行提取。萃取儀爐溫為100℃, 壓力為1500 psi, 提取劑為正己烷丙酮(4∶1, V/V)。萃取過程:加熱5 min, 靜態萃取5 min, 沖洗體積60%, 氮氣吹掃60 s, 循環2次。提取液用R210/R215型旋轉蒸發器(瑞士Buchi公司)濃縮至約1 mL。
PBDEs土壤提取液凈化:選取2.4節制備的10種SPE柱進行實驗, 實驗均設置6個平行, 并設置3個空白對照。凈化過程:5 mL正己烷活化SPE柱后, 將濃縮后的土壤提取液加入柱中, 用正己烷洗脫, 洗脫體積由洗脫曲線確定, 收集全部洗脫液, 濃縮定容至1 mL, GC測定。
2.6 ASE萃取溶劑優化
ASE萃取溶劑直接影響PBDEs的萃取效率及基質效應。為確定最佳萃取溶劑, 本實驗設計了以下4種萃取體系:正己烷、正己烷DCM(1∶1, V/V)、正己烷丙酮(4∶1, V/V)、正己烷丙酮(4∶1, V/V)進行萃取劑優化實驗, 采用酸性硅膠柱進行凈化, GC測定(具體步驟參照2.5節)。
3 結果與討論
3.1 方法線性關系和檢出限
由表1可知, 各目標化合物在各自濃度范圍內(10~500 ng/mL)均呈現出良好的線性關系, 相關系數均大于0.999, 檢出限為0.042~0.22 ng/mL。
3.2 SPE填料選擇和洗脫優化
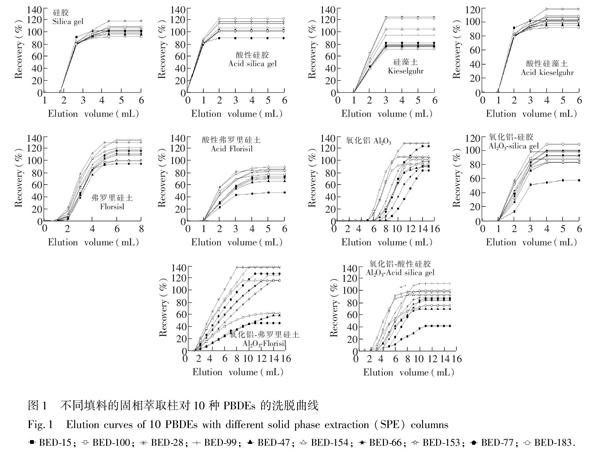
由圖1可知, 硅藻土柱、酸性硅藻土柱、弗羅里硅土柱、硅膠柱和酸性硅膠柱的洗脫趨勢大體一致, 對PBDEs的最大洗脫量出現在1~3 mL之間, PBDEs完全洗脫所需體積為5 mL(即洗脫體積, Vmax);酸性弗羅里硅土柱和氧化鋁硅膠復合柱洗脫趨勢大體一致, 最大洗脫量在2~5 mL之間, Vmax為8 mL; 氧化鋁柱、氧化鋁弗羅里硅土復合柱和氧化鋁酸性硅膠復合柱的最大洗脫量在5~10 mL之間, Vmax為15 mL;氧化鋁柱和氧化鋁酸性硅膠復合柱對PBDEs拖尾現象較明顯。
硅藻土具有良好的微孔結構, 比表面積較大, 吸附能力較強。實驗表明, 土壤提取液過硅藻土柱和酸性硅藻土柱后, 溶液顏色仍然較深, 說明兩種SPE柱除雜效果較差。由表 2可知, 雜質的存在影響了PBDEs的定量分析, 使PBDEs回收率偏高。
弗羅里硅土是一種極性較強的硅鎂型吸附劑, 對脂肪及類脂類雜質有較理想的去除效果, 常用于凈化土壤、植物和動物組織樣品的萃取液[20,29,30]。酸性弗羅里硅土極性更強, 同時, 濃H2SO4的存在可有效去除有色有機雜質。由表2可知, 兩種SPE柱的回收率相對偏低。
氧化鋁對目標物保留的主要機理是偶極偶極作用, 可用于除去土壤提取液中極性較強的有機酸類及其它極性雜質。氧化鋁對PBDEs有一定的保留能力, 這使得PBDEs在氧化鋁柱中出現不同程度的拖尾現象, Vmax為15 mL。隨著洗脫量增加, 雜質會隨洗脫液共流出, 凈化效果變差。為減少氧化鋁的拖尾現象, 本研究制備了氧化鋁硅膠柱、氧化鋁弗羅里硅土柱和氧化鋁酸性硅膠柱。實驗表明, 硅膠、酸性硅膠及弗羅里硅土中加入氧化鋁后, 洗脫速度明顯變慢, 洗脫時間明顯增加, 有機溶劑用量增多, 增加了環境污染。由表2可知, 氧化鋁柱和其它3種氧化鋁復合柱對PBDEs回收率在75.3%~110.9%之間。
硅膠表面由于吸水作用形成硅醇基, 合理數量的硅醇基可以增加硅膠與極性物質之間除疏水作用以外的氫鍵作用、離子相互作用和偶極偶極相互作用, 故硅膠表面硅醇基的數量決定了硅膠的吸附性能[31]。本研究對所用硅膠先進行去活化處理后, 再進行定量活化, 這樣制備出的硅膠的表面硅醇基含量均一, 性質穩定, 保證了實驗的重現性。酸改性使得硅膠表面引入了磺酸基, 增加了酸性硅膠對極性雜質吸附作用;同時濃H2SO4能較好地去除有色有機雜質[32,33]。由表2可知, 酸性硅膠柱與硅膠柱相比回收率更高, 除雜效果更好。由圖1還可知, 濃H2SO4改性的硅膠對PBDEs的作用機制及強度并無明顯變化, 而良好的回收率說明濃H2SO4的存在并沒有使PBDEs發生氧化降解等現象。
綜上, 對于土壤提取溶液的凈化, 當以正己烷作為洗脫液時, 酸性硅膠具有洗脫溶劑用量少、價格低廉、凈化效果好、回收率及重現性好等優點, 是一種理想的PBDEs土壤提取溶液SPE凈化的柱填料。
3.3 ASE萃取條件優化
由表 3可知, 對于土壤中PBDEs的提取, 正己烷回收率為87.6%~113.4%, 但提取穩定性(RSD=4.1%~9.1%)較正己烷丙酮(4∶1, V/V)體系差一些(RSD=1.7%~4.6%), 故極性和非極性溶劑的組合提取效果更好。在正己烷中加入極性溶劑(丙酮或二氯甲烷)后, PBDEs的提取效率增加, 而隨著提取體系極性增加, 所得提取溶液顏色越深, 提取出的雜質越多, 這增加了凈化過程的復雜性, 而未凈化除掉的雜質的存在會影響儀器的定性及定量準確性。
最終實驗選取正己烷丙酮(4∶1, V/V)作為ASE提取劑, 該提取體系對PBDEs的平均加標回收率為85.4%~103.1%;相對標準偏差為1.7%~4.6%, 實驗重現性較好;提取液經凈化后雜質較少, 且不影響定量分析, 可用作加速溶劑萃取土壤中PBDEs的提取劑。
3.4 方法準確度和精密度
以1.00 g海南磚紅壤作為基質, 分別加入濃度相當于10、40和100 ng/mL PBDEs進行加標回收實驗(參照2.5節), 每個濃度重復6次, 并設置3個空白對照。方法的準確度和精密度分別通過加標回收率和相對標準偏差表征。由表4可知, 低、中、高 3組(濃度分別為10, 40和100 ng/mL )的平均加標回收率分別為74.4%~115.2%, 87.5%~125.2%, 87.3%~115.9%;相對標準偏差分別為4.4%~14.4%, 5.0%~13.8%, 4.8%~7.1%。
3.5 實際樣品分析
采用上述優化后的方法, 對采集自某地的土樣進行分析。由表5可知, 該地區存在不同程度的PBDEs污染, ΣPBDEs為5.91~17.69 ng/g, 污染物以中、高溴代PBDEs為主。同時, 實際樣品分析結果說明, 優化的方法可用于測定土壤中的PBDEs。
4 結 論
采用ASE法提取土壤中的PBDEs, 正己烷丙酮(4∶1, V/V)的提取效果最佳;采用酸性硅膠SPE柱對樣品凈化, PBDEs完全流出僅需5 mL正己烷, 溶劑用量少, 環境污染小, 洗脫速度快, 雜質干擾少。本方法簡單、快捷, 具有良好的凈化效果、準確度和精密度(回收率74.4%~125.2%, RSD<15%), 良好的線性關系(r>0.999)及較低的檢出限(≤0.22 ng/mL), 可作為土壤介質中PBDEs的有效凈化和檢測方法。
References
1 Wenlu S, Ford J C, An L, Mills W J, Buckley D R, Rockne K J. Environ. Sci. Technol., 2004, 38(12): 3286-3293
2 Gouin T, Harner T. Environ. Int., 2003, 29(6): 717-724
3 Brouwer A, Morse D C, Lans M C, Schuur A G, Murk A J, KlassonWehler E, Bergman A, Visser T J. Toxicol. Ind. Health, 1998, 14(12): 59-84
4 Glinoer D. Endocr. Rev., 1997, 18(3): 404-433
5 Eriksson P. Neurotoxicology, 1997, 18(3): 719-26
6 Costa L G, Giordano G. Neurotoxicology, 2007, 28(6): 1047-1067
7 Skarman E, Darnerud P O, hrvik H, Oskarsson A. Environ. Toxicol. Pharmacol., 2005, 19(2): 273-281
8 Branchi I, Bichler Z, BergerSweeney J, Ricceri L. Neurosci. Biobehav. Rev., 2003, 27(12): 141-153
9 Per Ola D, Sofia R. Chemosphere, 2006, 62(3): 485-493
10 Elliott J E, Wilson L K, Wakeford B. Environ. Sci. Technol., 2005, 39(15): 5584-91
11 Pu W, Zhang Q, Wang Y, Wang T, Li X, Lei D. Jiang G. Anal. Chim. Acta, 2010, 663(1): 43-48
12 LU Min, HAN ShuYuan, YU YingXin, ZHANG DongPing, WU MingHong, SHENG GuoYing, FU JiaMo. Journal of Instrumental Analysis, 2009, 28(1): 1-6
陸 敏, 韓姝媛, 余應新, 張東平, 吳明紅, 盛國英, 傅家謨. 分析測試學報, 2009, 28(1): 1-6
13 Abrha Y, Raghavan D. J. Hazard. Mater., 2000, 80(13): 147-157
14 JIANG JinHua, CHEN Tao. Chinese J. Anal. Chem., 2009, 37(11): 1627-1632
江錦花, 陳 濤. 分析化學, 2009, 37(11): 1627-1632
15 Tao S, Guo L Q, Wang X J, Liu W X, Ju T Z, Dawson R, Cao J, Xu F L, Li B G. Sci. Total Environ., 2004, 320(1): 1-9
16 Tapie N, Le Menach K, Pasquaud S, Elie P, Devier M H, Budzinski H. Chemosphere, 2011, 83(2): 175-185
17 Liaud C, Millet M, Le Calvé S. Talanta, 2015, 131: 386-394
18 Yus V, Quintas G, Pardo O, Pastor A, Guardia M D L. Talanta, 2006, 69(4): 807-815
19 XU NengBin, QIAN FeiZhong, FENG JiaYong, WANG ShengLe, HONG ZhengFang, XU LiHong, CHEN ZhongQuan. Chinese J. Anal. Chem., 2015, 43(2): 251-256
徐能斌, 錢飛中, 馮加永, 汪晟樂, 洪正昉, 徐立紅, 陳鐘佺. 分析化學, 2015, 43(2): 251-256
20 Król S, Zabiegaa B, Namies'nik J. Talanta, 2012, 93: 1-17
21 FernándezPeralbo M, Vera C F, PriegoCapote F, de Castro M L. Talanta, 2014, 126: 170-176
22 Willenberg I, Von Elsner L, Steinberg P, Schebb N H. Food Chem., 2015, 166: 537-543
23 Liu Y, Nielsen M, Staerk D, Jger A K. J. Ethnopharmacol., 2014, 155(2): 1276-1283
24 Rossmann J, Schubert S, Gurke R, Oertel R, Kirch W. J. Chromatogr. B, 2014, 969: 162-170
25 Wang X, Li P. Food Chem., 2015, 173: 897-904
26 Wiese S, Wubshet S G, Nielsen J, Staerk D. Food Chem., 2013, 141(4): 4010-4018
27 Heuett N V, Ramirez C E, Fernandez A, Gardinali P R. Sci. Total Environ., 2015, 511: 319-330
28 Roszko M, Szymczy K, Jedrzejczak R. Anal. Chim. Acta, 2013, 799(17): 88-98
29 Sun J, Liu J, Liu Q, Qu G, Ruan T,Jiang G. Talanta, 2012, 88,(1): 669-676
30 Liu H, Zhang Q, Cai Z, Li A, Wang Y, Jiang G. Anal. Chim. Acta, 2006, 557: 314-320
31 YANG XinLi, WANG JunDe, XIONG BoHui. Chinese Journal of Chromatography, 2000, 4(18): 308-312
楊新立, 王俊德, 熊博暉. 色譜, 2000, 4(18): 308-312
32 Manirakiza P, Covaci A, Nizigiymana L, Ntakimazi G, Schepens P. Environ. Pollut., 2002, 117(3): 447-455
33 Lino C M, Silveira M I N D. J. Chromatogr. A, 1997, 769(97): 275-283
Abstract A method was established for the determination of 10 polybrominated diphenyl ethers (PBDEs) in soil using accelerated solvent extraction (ASE), solid phase extraction (SPE) and gas chromatography with electron capture detector (GCECD). ASE was used to obtain 10 PBDEs in soil and a good extraction system was investigated by comparison of kinds of extraction systems (nhexane, nhexane∶acetone (4∶1, V/V), nhexane∶acetone (1∶1, V/V), and nhexane∶dichloromethane (1∶1, V/V)). Besides, 10 kinds of SPE columns with different filters were applied to the purification of 10 PBDEs in soil solution. The optimized conditions were acquired by using the mixture of nhexane and acetone (4∶1, V/V) as extract and acidic silica gel column for the purification. Under the optimized conditions, the average recoveries of 10 PBDEs in soil ranged from 74.4% to 125.2% with the RSDs of 4.4%-14.4%. Method detection limits (MDLs) were 0.04-0.22 ng/mL. The method is simple, rapid and efficient, which has been successfully applied to determine 10 PBDEs in contaminated soil.
Keywords Soil; Polybrominated diphenyl ethers; Accelerated solvent extraction; Solid phase extraction; Gas chromatography
猜你喜歡
中國纖檢(2016年12期)2017-01-20 09:28:19
現代農業科技(2016年20期)2016-12-20 14:51:09
現代農業科技(2016年20期)2016-12-20 09:05:36
分析化學(2016年7期)2016-12-08 00:09:44
分析化學(2016年7期)2016-12-08 00:07:08
價值工程(2016年29期)2016-11-14 01:34:54
科技視界(2016年24期)2016-10-11 18:58:00
考試周刊(2016年39期)2016-06-12 16:01:44
中國科技博覽(2016年4期)2016-04-25 07:25:47
中國科技博覽(2016年8期)2016-04-25 04:57:50
