MKP1通過抑制JNK信號途徑減輕淀粉樣蛋白的神經毒性作用
2017-08-15 00:46:28王婷婷侯瑋琛鄭勝哲
中風與神經疾病雜志 2017年12期
孫 晶, 范 佳, 王婷婷, 侯瑋琛, 鄭勝哲
AD的病理特征為神經元數量減少、細胞內神經原纖維纏結和細胞外β樣淀粉蛋白(amyloid beta,Aβ)沉積[1]。在疾病早期,AD患者腦組織內即可見到聚集狀的 Aβ[2],通過調節下游信號途徑如絲裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)等誘導神經炎癥和氧化應激反應,并促進神經元凋亡,產生的級聯反應推動著AD病變的發展[3,4]。然而,Aβ調控MAPK信號通路的具體分子機制尚未完全闡明。MKP1是絲裂原活化蛋白激酶磷酸酶(MKPs,mitogen-activated protein kinase phosphatases)家族中最重要的一員,是p38、JNK1/2和ERK1/2負調控的主要因子[5]。MKP1被報道在多種疾病發生發展中通過調控MAPK信號途徑參與氧化應激、炎癥反應和細胞凋亡[6,7],但MKP1/MAPK信號軸對Aβ所致氧化應激和炎癥反應的調控作用還未完全清楚。在本研究中。因此,本研究對MKP1在Aβ所致MAPK激活、神經炎癥和細胞凋亡中的調控作用作初步探索,旨在為AD開發新的有潛力的臨床治療手段提供理論依據。
1 材料與方法
1.1 細胞培養 野生型PC12細胞、過表達MKP1和穩定敲除MKP1的PC12細胞培養在含10%胎牛血清的DMEM培養基中,置于5% CO2,37 ℃培養箱中,每3 d換液一次。
1.2 實驗試劑 CCK-8試劑盒購自于Sigma公司,過氧化物歧化酶(SOD)和丙二醛(MDA)試劑盒購自于南京建成生物科技公司。MKP1和JNK抗體購自于Abcam公司。逆轉錄和PCR試劑盒購自于全式金公司。
1.3 實驗分組 在細胞培養基中加入不同濃度的Aβ42(0 μmol,0.1 μmol,1 μmol,10 μmol 和100 μmol,每組6孔),處理24 h后CCK-8檢測細胞活性。向細胞培養基中加入10 μmol 的Aβ42,在不同時間點(0 h、6 h、12 h、18 h和24 h,每組6孔),通過qRT-PCR和Western blot檢測MKP1表達。將野生型PC12細胞分為對照組(Control)和Aβ42組,敲除MKP1的細胞為MKP1 KD+Aβ42組,過表達MKP1細胞為MKP1+Aβ組,每組6孔,后3組加入10 μmol Aβ42,置于培養箱中24 h后進行氧化應激和炎癥反應的檢測。
1.4 CCK-8檢測 為檢測各組細胞的細胞活性,按照說明書對各組細胞進行CCK-8檢測,檢測過程簡略如下:將細胞接種到96孔板中(4000/well),按不同濃度(0、0.1、1、10、100 μmol )Aβ處理后,更換培養基。每孔加入10 μl CCK8。繼續培養2 h。于酶標儀下測定450 nm吸光度。
1.5 qRT-PCR 按照說明書方法通過Trizol提取總RNA,使用Superscript III Reverse Transcriptase試劑盒進行cDNA合成。將2.5 μl合成的cDNA、1 μl特異性引物與qPCR試劑充分混合后,在ABI 7500 PCR儀上進行實時定量PCR測定,以GAPDH mRNA內參,定量產物濃度。
1.6 Western blot檢測 提取細胞總蛋白,測定總蛋白濃度。每個樣品取總蛋白15 μg與4 μl 6×loading buffer混合,行10% SDS-PAGE不連續凝膠電泳,濕法電轉膜,封閉2 h,洗脫后加一抗1∶1000于4 ℃孵育過夜,TBST洗脫,再加入辣根過氧化物酶標記的二抗,室溫孵育2 h后行ECL化學發光法顯示結果。
1.7 DCFH-DA檢測 為檢測細胞內ROS水平,將2×105PC12細胞種于6孔板,按照說明書加入終濃度為 20 nmol/L的 DCFH-DA,于5% CO2,90%濕度,37 ℃培養箱中孵育30 min。在熒光顯微鏡下觀察綠色熒光表達情況并拍照。
1.8 線粒體膜電位(ΔΨm)檢測 將PC12細胞按照2×105個/ml接種于6孔板,按照上述分組方法給予處理4組細胞。各組加入等量DMSO溶解的羅丹明123終濃度為5 μmol/L的Fluo-3/AM Ester和0.0625%重量比的Pluronic F127,于5%CO2、90%濕度、37 ℃培養箱中孵育45 min。PBS洗兩次后,1500 rpm離心5 min,在激發波長為485 nm時流式細胞儀檢測各組熒光強度(MFI)。
1.9 化學發光法檢測 嚴格按照說明書對細胞內SOD活性和MDA水平行進檢測:細胞裂解后,12000 g離心,取上清液加入檢測液,95 ℃恒溫水浴40 min,冷卻后4000 g離心10 min。提取上清于波長532 nm,光徑1 cm處測定并記錄OD值。
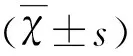
2 結 果
2.1 Aβ42下調PC12細胞中MKP1的表達 隨濃度的增大,Aβ42對PC12的毒性增大,當Aβ42濃度大于10 μmol/L時,PC12細胞的活性受到明顯的抑制(P<0.01),可使PC12細胞活性下降至正常細胞的50%。qRT-PCR結果顯示,10 μmol/L Aβ42刺激6 h,即可觀察到MKP1 mRNA的下降(P<0.05),在刺激12 h時達到最底峰,約為正常水平的25%(P<0.01)。Western blot結果顯示,MKP1蛋白表達在Aβ42刺激12 h后出現明顯下調(P<0.01),在18 h時下調最為明顯,約為正常MKP1表達水平的30%。
2.2 MKP1調控Aβ42所致的氧化應激反應 Aβ42可導致PC12細胞中大量ROS生成,在熒光顯微鏡下呈綠色熒光。敲除MKP1后,PC12細胞中熒光強度明顯高于Aβ42組,而過表達MKP1可減輕PC12細胞中的熒光強度。通過流式細胞術檢測我們可以發現Aβ42可下調PC12細胞的ΔΨm(P<0.05),而過表達MKP1可恢復PC12細胞的ΔΨm水平(P<0.05)。敲除MKP1會下調PC12細胞中SOD活性(P<0.01),但并不會增加MDA水平(P>0.05)。而過表達MKP1可上調SOD活性(P<0.05),并下調MDA水平(P<0.01)。
2.3 MKP1調控Aβ42所致的神經炎癥 Aβ42刺激可上調p-JNK含量,敲除MKP1可進一步增加這種作用(P<0.05),而過表達MKP1可減輕這種作用(P<0.05)。通過qRT-PCR對PC12細胞中炎癥因子TNF-α和IL-1β mRNA水平進行檢測,結果顯示敲除MKP1可加重Aβ42刺激對炎癥因子TNF-α和IL-1β mRNA表達的上調作用(P<0.05),而過表達MKP1可減輕Aβ42刺激所引起的炎癥因子釋放(P<0.05)。
3 討 論
在AD早期,Aβ在腦組織中聚集,可通過激活MAPK下游信號通路誘導大量ROS生成以及強烈持續的炎癥反應,這最終導致了神經細胞的凋亡[8]。這些現象很早即在AD動物模型和體外實驗中被驗證,然而Aβ是如何激活MAPK信號途徑,我們目前所知還很少。在本研究中,我們發現Aβ42對PC12細胞的毒性作用隨濃度的增大而增大,當Aβ42濃度為10 μmol/L時,PC12細胞活性下降至正常細胞的50%,因此我們選擇該濃度觀察Aβ42對PC12細胞中MKP1表達的影響。我們的結果顯示,Aβ42刺激6 h時即可觀察到MKP1 mRNA的下降,但蛋白表達在12 h左右才發生明顯變化。MKP1 mRNA在刺激12 h時達到最底峰,而其蛋白表達在18 h時才出現該種現象。MKP1在Huntington’s disease患者腦組織內表達下調[9],但目前尚無AD疾病中MKP1表達變化的數據。我們的結果初步顯示,至少在體外MKP1在Aβ的作用下是表達下調的,提示MKP1可能參與到Aβ致病的機制中。
大量證據表明,ROS生成過多伴隨抗氧化能力的相對不足導致的氧化應激反應在AD中扮演著重要的角色[10,11]。在本研究中,通過DCFH-DA檢測發現,經Aβ42刺激之后PC12細胞內綠色熒光DCF表達增強,表明Aβ可誘導神經細胞中ROS的大量生成。敲除MKP1后,PC12細胞中ROS生成明顯增多,而過表達MKP1可減輕PC12細胞中ROS含量。ΔΨm是反應氧化應激的一個生物學指標,ROS可導致其ΔΨm的降低[12]。敲除MKP1或過表達MKP1同樣會相應下調或上調Aβ42引起的線粒體膜電位損害效應。并且,敲除MKP1會下調PC12細胞中SOD的活性,而過表達MKP1可上調SOD活性并下調MDA水平。此結果表明MKP1具有調節Aβ所致神經細胞內氧化應激的作用。JNK是MAPK通路的一重要分支,在炎癥發生過程中起重要作用。在本研究中,我們發現經過Aβ42刺激后,細胞內p-JNK含量增多,表明Aβ可促進JNK的活化。而在敲除或過表達MKP1的細胞中的結果表明,MKP1可以調控由Aβ誘導的JNK信號通路的激活,并可以減輕炎癥因子TNF-α和IL-1β的生成。
綜上,我們的研究結果表明,Aβ刺激可下調MKP1表達,而MKP1可通過下調JNK信號通路調控Aβ所致的氧化應激和炎癥反應。本研究不僅解釋了Aβ激活MAPK信號途徑的機制,還為靶向作用于JNK信號軸的AD治療藥物的研發提供了一條新途徑。
[1]Goedert M.Neurodegeneration.Alzheimer’s and Parkinson’s diseases:The prion concept in relation to assembled Abeta,tau,and alpha-synuclein[J].Science,2015,349(6248):1255555.
[2]Puzzo D,Gulisano W,Arancio O,et al.The keystone of Alzheimer pathogenesis might be sought in Abeta physiology[J].Neuroscience,2015,307:26-36.
[3]Petersen RB,Nunomura A,Lee HG,et al.Signal transduction cascades associated with oxidative stress in Alzheimer’s disease[J].J Alzheimers Dis,2007,11(2):143-152.
[4]Smith WW,Gorospe M,Kusiak JW.Signaling mechanisms underlying Abeta toxicity:potential therapeutic targets for Alzheimer’s disease[J].CNS Neurol Disord Drug Targets,2006,5(3):355-361.
[5]Chi H,Flavell RA.Acetylation of MKP-1 and the control of inflammation[J].Sci Signal,2008,1(41):44.
[6]Ma G,Pan Y,Zhou C,et al.Mitogen-activated protein kinase phosphatase 1 is involved in tamoxifen resistance in MCF7 cells [J].Oncol Rep,2015,34(5):2423-2430.
[7]Wancket LM,Frazier WJ,Liu Y.Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology,physiology,and disease[J].Life Sci,2012,90(7/8):237-248.
[8]Origlia N,Arancio O,Domenici L,et al.MAPK,beta-amyloid and synaptic dysfunction:the role of RAGE[J].Expert Rev Neurother,2009,9(11):1635-1645.
[9]Taylor DM,Moser R,Regulier E,et al.MAP kinase phosphatase 1 (MKP-1/DUSP1) is neuroprotective in Huntington’s disease via additive effects of JNK and p38 inhibition[J].J Neurosci,2013,33(6):2313-2325.
[10]Luque-Contreras D,Carvajal K,Toral-Rios D,et al.Oxidative stress and metabolic syndrome:cause or consequence of Alzheimer’s disease[J].Oxid Med Cell Longev,2014,2014:497802.
[11]Aliev G,Priyadarshini M,Reddy VP,et al.Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease[J].Curr Med Chem,2014,21(19):2208-2217.
[12]Vayssier-Taussat M,Kreps SE,Adrie C,et al.Mitochondrial membrane potential:a novel biomarker of oxidative environmental stress[J].Environ Health Perspect,2002,110(3):301-305.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
世界科學技術-中醫藥現代化(2020年2期)2020-07-25 02:05:56
電子制作(2018年11期)2018-08-04 03:25:42
海峽科技與產業(2016年3期)2016-05-17 04:32:12
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
西南軍醫(2016年6期)2016-01-23 02:21:19
