2例脊髓小腦性共濟失調患者的臨床特點及基因測定分析
2017-11-15 14:25:50陳成芳施俊峰王亮亮李杰馬穎王國祥湯武裝趙彥平
中外醫學研究 2017年26期
陳成芳 施俊峰 王亮亮 李杰 馬穎 王國祥 湯武裝 趙彥平
【摘要】 目的:探討2例脊髓小腦性共濟失調患者的臨床特點及基因突變特點。方法:詳細收集2例住院患者臨床病史資料,并對其進行基因檢測,采用聚合酶鏈反應分別擴增SCA1、SCA2、SCA3、SCA6、SCA7、SCA12及DRPLA致病基因的CAG重復序列。結果:1例女性患者診斷為SCA1,另1例男性患者診斷為SCA2。結論:脊髓小腦性共濟失調患者的臨床表現存在一定差異,檢測基因的CAG重復數是確診的有效方法。
【關鍵詞】 脊髓小腦性共濟失調; 基因; 臨床特點
doi:10.14033/j.cnki.cfmr.2017.26.086 文獻標識碼 B 文章編號 1674-6805(2017)26-0163-02
Clinical Features and Genetic Analysis of two Patients with Spinocerebellar Ataxia/CHEN Cheng-fang,SHI Jun-feng,WANG Liang-liang,et al.//Chinese and Foreign Medical Research,2017,15(26):163-164
【Abstract】 Objective:To investigate the clinical characteristics and gene mutation of 2 patients with spinocerebellar ataxia.Method:The clinical data of 2 inpatients were collected and analyzed by polymerase chain reaction(PCR).The CAG repeats of SCA1,SCA2,SCA3,SCA6,SCA7,SCA12 and DRPLA were amplified by polymerase chain reaction.Result:SCA1 was diagnosed in one female patient and SCA2 in another male patient.Conclusion:The clinical manifestations of patients with spinocerebellar ataxia are different,and the number of CAG repeats of the test gene is an effective method for diagnosis.
【Key words】 Spinocerebellar ataxia; Genes; Clinical features
First-authors address:Peoples Hospital of Yixing,Yixing 214200,China
脊髓小腦性共濟失調(spinocerebellar ataxia,SCAs)是以小腦性共濟失調為主要臨床癥狀(發音含糊、步態不穩、吞咽困難、肢體乏力甚至失去意識)的常染色體顯性遺傳性疾病,其病因主要是基因中有段異常的CAG核酸重復序列發生倍增突變產生多聚谷氨酰胺[1],基因檢測是目前確診SCAs的有效方法,目前尚無特效治療方法。
1 病例介紹
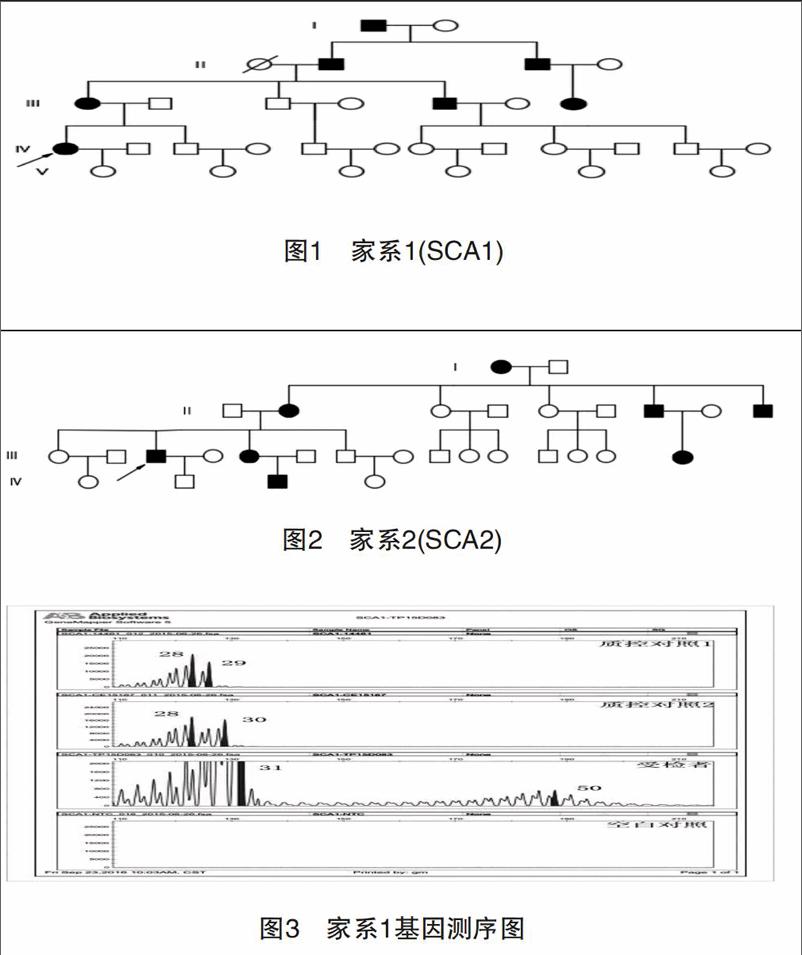
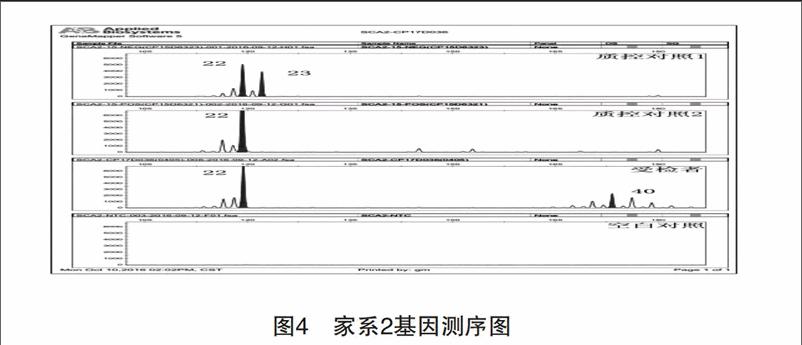
收集2例診斷明確的脊髓遺傳型共濟失調患者,取得患者知情同意后,病史資料和外周靜脈血用于研究分析。病例1,女性,44歲,病程1年,臨床表現:步態不穩,共濟失調,吞咽困難、腱反射活躍。追蹤家系發現5代7例臨床表現相似患者(圖1)。病例2,男性,49歲,病程16年,臨床表現:進行性步態不穩,枕部疼痛、顱內壓增高、吞咽困難、病理性眼球震顫、呼吸困難等,膜反射減弱,巴彬斯基征陽性,同時出現四肢間歇性痙攣,疼痛。追蹤家系發現4代8例臨床表現相似患者(圖2)。然2組家系其他發病者均已亡故,而現存成員拒絕基因檢測,未能完全確診。
2 基因檢測結果
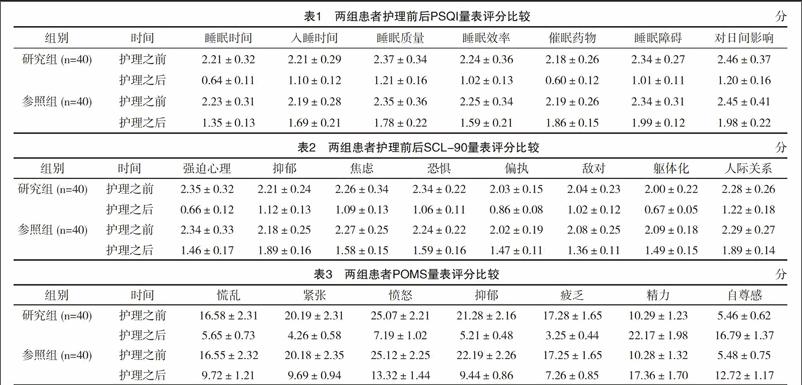
靜脈血送廣州市國際生物島基因測序,采用聚合酶鏈反應分別擴增SCA1、SCA2、SCA3、SCA6、SCA7、SCA12及DRPLA相關基因的CAG重復序列。對2例患者進行檢測,病例1患者,SCA1基因一個等位基因重復數為31次,屬于正常范圍,另一個為50次,屬于全突變范圍(圖3)。其他SCA2、SCA3、SCA6、SCA7、SCA12及DRPLA相關基因的重復數均屬正常范圍;病例2患者SCA2基因一個等位基因重復數為22次,屬于正常范圍,另一個為40次,屬于全突變范圍(圖4),另外SCA1、SCA3、SCA6、SCA7、SCA12及DRPLA相關基因的重復數均屬正常范圍。
3 討論
脊髓小腦性共濟失調是基因中異常的CAG核酸重復序列發生倍增突變引起的,臨床以發音含糊、步態不穩、吞咽困難等為主要表現的,具有遺傳性的單基因神經系統變性疾病[2]。SCA的病變部位主要為小腦、腦干和脊髓[3]。隨著醫學遺傳學的發展,人們發現,不同基因分型SCA其臨床其臨床癥狀和遺傳差異存在高度的異質性[4]。目前發現的SCA致病基因亞型主要有27個[5]。不同的國家和種族,SCA各亞型的分布不同,SCA3是我國脊髓小腦性共濟失調患者基因檢出率最高的一個亞型,約占SCA患者的50%以上,但對于SCA1、SCA2兩個亞型國內報道并不多[6]。
本研究發現,SCA1主要的臨床表現為構音障礙、書寫困難、肢體共濟失調,通常也表現為眼球震顫、掃視異常。隨著病情進展,還可出現視神經萎縮、癡呆和不同程度的精神障礙[7]。SCA1在正常等位基因中含有6~44個CAG重復片段。超過20個重復片段可被CAT片段打斷[8],異常等位基因CAG重復片段39個,且不能被CAT打斷,DNA分析證明SCA1基因位于染色體6P23。SCA2主要表現為軀體共濟失調、構音障礙、遲發型慢眼動。部分患者可能出現眼肌麻痹和舞蹈癥,沒有明顯的臨床癥狀能與SCA1型區分。Gispert等[9]將SCA2基因突變定位于12P24,正常等位基因CAG重復數為15~24,CAG重復在35次以上且序列中沒有CAA插入便會引起臨床癥狀。兩例患者均通過基因檢測符合突變范圍。患者SCA2盡管擴增數目不大,但患者臨床癥狀卻很嚴重,考慮與文獻[10]報道的基因突變純合子易加重患者臨床癥狀相關。研究報道SCAs核苷酸實變擴展的數目與年齡呈負相關,與癥狀嚴重程度呈正相關[11-12],在SCA3中這種相關關系最顯著,SCA1、2目前還沒有大規模的報道。endprint
本研究患者分別為SCA1、SCA2兩個亞型,這兩個亞型在我國相對較少見,本研究病例1為SCA1型,其癥狀為步態不穩、共濟失調、吞咽困難、腱反射活躍,與國外對于該亞型癥狀的報道基本相似。國外研究在SCA1、SCA2存在遺傳早現現象,本研究結果未發現該現象,考慮與本研究納入病例數較少有關。
據此筆者明確了SCA的一般診斷思路:首先根據患者的臨床表現基本確定為SCA,后收集家族病史;排除腦血管、腫瘤等非遺傳性病因;通過生化診斷確定某些特殊的生化異常遺傳性共濟失調。對于染色體顯性的共濟失調,根據我國不同基因亞型發病率從高到低進行檢測,首先檢測SCA3,如為陰性再檢測SCA1、SCA2。SCA1、SCA2兩例患者臨床特點與SCA系列疾病癥狀相似,無特異性表現,僅憑臨床癥狀及影像學只能做臨床診斷,很難對其分型,通過基因手段是確診的唯一辦法。
分析本研究的不足,由于本研究報道病例數相對較少,未發現SCA家系遺傳早現現象,即發病年齡一代比一代逐漸提前,病情程度一代比一代逐漸加重,對于年齡和CAG擴增數之間的關系,還需通過基因測序發現更多病例進一步證實。
參考文獻
[1] Shakkottai V G,Fogel B L.Clinical neurogenetics autosomal dominant spinocerebellar ataxia[J].Neurol Clin,2013,31(4):987-1007.
[2]吳丹丹.脊髓小腦性共濟失調兩個家系的分子遺傳學研究[D].北京:中國人民解放軍軍事醫學科學院,2016.
[3]魏笑凡.脊髓小腦性共濟失調3型一家系的臨床及電生理特征分析[D].北京:協和臨床醫學院,2014.
[4]張思仲.加強醫學遺傳學的轉化,促進我國臨床遺傳學的發展[J].中華醫學遺傳學雜志,2012,29(6):728-730.
[5]李佳.脊髓小腦共濟失調致病基因X在果繩中的異常表型探究[D].長沙:中南大學,2014.
[6]吳志英.脊髓小腦性共濟失調3型的臨床異質性及其在我國高發的原因分析[A].2015年浙江省神經病學學術年會論文匯編[C].2015.
[7] Schols L,Bauer P,Schmidt T,et al.Autosomal ataxias an update[J].Curr Opin Neurol,2007,20(4):438-446.
[8] Chung M Y,Ranum L P,Duvick L A,et al.Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellay ataxia type Ⅰ[J].Nat Genet,1993,5(3):254-258.
[9] Gispert S,Twells R,Orozco G,et al.Chromosomal assognment of the second locus for autosomal dominant cerebellar ataxia (SCA2)to chromosome 12q23-24.1[J].Nat Genet,1993,4(3):295-299.
[10] ZENG S,ZENG J,HEM,et al.Chinese homozyqous Machado-Jo-seph disease (MJD)/SCA3:a case report[J].J Hum Genet,2015,60(3):157-160.
[11] Fujigasaki H,Verma I C,Camuzat A,et al.SCA12 is a rarelocus for autosoma dominant eerehellar ataxia a study ot an Indian family[J].Ann Neurol,2001,49(1):117-121.
[12] Bahl S,Virdi K,Mittal U,et al.Evidence of a common founder for SCA12 in the Indian poputation[J].Ann Hum Genet,2005,69(Pt5):528-534.
(收稿日期:2017-05-14)endprint
