響應曲面-多指標評價法優(yōu)化枳子提取工藝*
2018-05-16 02:08:12蔡向杰屈云萍王亞濤姜國志
西部中醫(yī)藥 2018年4期
蔡向杰,屈云萍,王亞濤,李 哲,姜國志
1神威藥業(yè)集團有限公司,河北 石家莊 051400;2中藥注射劑新藥技術開發(fā)國家地方聯合工程實驗室
枳椇為鼠李科(Rhamnaceae)拐棗屬(Hovenia Thunb.)落葉喬木,始載于《唐本草》,又名拐棗、金鉤子、甜半夜等。枳椇子系枳椇植物干燥成熟的種子,有清熱利尿,解酒毒之功效,主治煩熱、口渴、嘔吐、二便不利等癥。枳椇子應用歷史悠久,是國家衛(wèi)生部批準的藥食同源植物之一,其主要化學成分包括:皂苷、糖苷、生物堿、黃酮類化合物、脂肪酸及其他微量成分[1-4]。許多學者研究認為枳椇子的療效與其所含成分的抗氧化作用密切相關,其中以黃酮類和皂苷類物質尤為重要。枳椇子作為藥食同源的中藥之一,具有良好的發(fā)展前景,其有效成分的提取和純化對枳椇子的質量控制意義重大。響應面分析法(response surface methodology,RSM)是利用合理的設計并通過試驗得到一定數據,采用多元二次逐步回歸方差擬合因素與響應值之間的函數關系,通過回歸方程分析尋找最佳工藝參數,解決多變量問題的一種統(tǒng)計方法[5-10],已經廣泛應用于中藥材提取工藝研究中[11-13]。實驗以指標成分(皂苷含量、總黃酮含量及干膏收率)的綜合得分為指標,采用響應曲面法建立多元二次回歸方程,用以優(yōu)化枳椇子的提取工藝,為其開發(fā)和應用及質量控制提供數據來源,為其產業(yè)化提取數據支持。
1 材料與方法
1.1 試藥 枳椇子藥材購于安國市振宇中藥材飲片有限公司,產地湖南,批號:150608。經河北省藥品檢驗研究院孫寶惠老師檢驗鑒定為鼠李科(Rhamnaceae)拐棗屬(Hovenia Thunb.)枳椇藥材的種子。蘆丁對照品(純度92.5%)購自中國藥品生物制品檢定所,規(guī)格100 mg/支,批號:100080-200707。乙醇、乙醚、正丁醇等其他試劑均為分析純。
1.2 儀器 METTLER-ALC-210.4電子分析天平(上海梅特勒托利多儀器有限公司);HC-400Y2粉碎機;紫外可見分光光度計;HH-SB型電熱恒溫水浴鍋(科偉儀器);SHB-Ⅲ循環(huán)水式真空泵(鄭州長城科工貿有限公司)。
1.3 方法
1.3.1 樣品處理 將枳椇子用粉碎機粉碎至40~60目,混勻密封備用。
1.3.2 皂苷含量和總黃酮含量的提取 精密稱定一定量的枳椇子粉末,加入一定溶劑配成料液,在一定溫度下提取一定的時間,提取液過濾濃縮成干膏粉。
1.3.3 總黃酮的含量測定[14]精密稱取120℃干燥至恒重的蘆丁對照品50 mg,置于100 mL量瓶中,用80%乙醇溶解并定容。精密量取上述對照品溶 液 0.0、1.0、2.0、3.0、4.0、5.0 mL 分 別 置 入25 mL量瓶中,加5%NaNO2溶液1.0 mL,搖勻,放置6分鐘,加10%Al(NO3)3溶液1.0 mL,搖勻;放置6分鐘,加入4%NaOH溶液10.0 mL,用80%乙醇稀釋至刻度,混勻。放置15分鐘在500 nm波長處,以相應試劑作空白,分別測吸收度。以吸收度A為縱坐標,濃度C為橫坐標,繪制標準曲線。
1.3.4皂苷的含量測定[15]取枳椇子干膏粉約0.5 g,精密稱定,用水20 mL分次溶解并洗入分液漏斗中。用乙醚分次振搖脫脂,每次20 mL,至乙醚層無色,棄去乙醚洗液。用水飽和正丁醇分次振搖提取,每次20 mL,至正丁醇層無色,合并正丁醇提取液于已知重量蒸發(fā)皿中,置水浴上蒸干,105℃干燥至恒重,稱重,計算。

1.4 單因素實驗
1.4.1 提取時間對結果的影響 稱取枳椇子粉末1.0 g,以純化水作為溶劑提取1次,分別提取1、2、3、4、5、6 h,按照“1.3.3 項”及“1.3.4 項”的方法進行測定。
1.4.2 提取次數對結果的影響 稱取枳椇子粉末 1.0 g,以純化水作為溶劑,分別提取 1、2、3、4、5、6次,每次提取 1 h,按照“1.3.3項”及“1.3.4項”的方法進行測定。
1.4.3 溶劑倍量對結果的影響 稱取枳椇子粉末 1.0 g,分別加入 5、6、7、8、9、10 倍量純化水提取1次,提取2小時,按照“1.3.3項”及“1.3.4項”的方法進行測定。
1.5 響應面分析因素水平的選取和設計 通過單因素實驗結果篩選出顯著性影響因素后,以顯著性影響因素(提取時間、提取次數、溶劑倍量)作為考察因子,優(yōu)化枳椇子的提取工藝。響應曲面因素水平編碼見表1。

表1 響應曲面實驗因素水平編碼
2 結果與分析
2.1 綜合評價指標和權重的選擇 以提取時間、提取次數、溶劑倍量為因素,每個因素選3個水平,以干膏收率與干膏中總黃酮含量、皂苷含量為指標,根據指標間的重要性差異給出各指標的權重,確定綜合評分=干膏收率×30+干膏中總黃酮含量×40+ 干膏中皂苷含量×30[16]。
2.2 提取溶劑的選擇[17-18]目前文獻中報道,枳椇子提取方式有水提和70%醇提。所以本實驗分別加入5倍的純化水和70%乙醇,提取2次,每次2小時,旨在對枳椇子提取方式進行考察分析。結果表明,水提皂苷含量為4.15%,總黃酮含量為4.26%,收率為6.86%。而醇提皂苷含量為3.57%,總黃酮含量為3.88%,收率為4.97%。按2.1進行綜合評分后結果為:水提綜合評分為5.007,而醇提綜合評分為4.114。可見水提優(yōu)于醇提,所以提取溶劑最終確定為純化水。
2.3 繪制總黃酮標準曲線 按照1.3.3繪制總黃酮標準曲線,線性回歸方程為A=0.038 65C-0.00119,R=0.9999,線性范圍 5.11~25.55mg/L。2.4 單因素實驗
2.4.1 提取時間對結果的影響 稱取枳椇子粉末1.0 g,以純化水作為溶劑提取1次,分別提取1、2、3、4、5、6 次,按照“1.3.3 項”及“1.3.4 項”的方法進行測定,并根據2.1中方法對結果進行綜合評分。提取時間為1~3小時隨著提取時間的延長,得分逐漸增加,當提取時間達到3 h時得分最高。繼續(xù)提取,綜合得分反而下降,并在提取5 h時趨于平穩(wěn)。原因是隨著提取時間的延長,產生了大量雜質,使得收率雖有所增加,但2個含量均大大下降使得分下降。因此,綜合考慮將提取時間定為3小時最合適。見圖1。
2.4.2 提取次數對結果的影響 稱取枳椇子粉末 1.0 g,以純化水作為溶劑,分別提取 1、2、3、4、5、6次,每次提取 1 小時,按照“1.3.3項”及“1.3.4項”的方法進行測定,并通過“2.1項”對結果進行綜合評分。提取次數為1~3次時隨著提取次數的增加,得分逐漸增加。增加提取次數得分反而下降,并趨于平穩(wěn)。原因是提取4次時已經將有效成分完全提取,再增加提取次數導致提取出雜質,影響了綜合得分,所以將提取次數定為3次最為合適。見圖2。
2.4.3 溶劑倍量對結果的影響 稱取枳椇子粉末 1.0 g,分別加入 5、6、7、8、9、10 倍量純化水提取 1次,提取 2小時,按照“1.3.3項”及“1.3.4項”的方法進行測定,并通過“2.1項”對結果進行綜合評分。溶劑倍數5~8倍時得分逐漸增加,當溶劑量為8倍時得分最高,這是因為提取溶劑的增加使藥材充分的浸提,有利于有效成分的浸出,但當溶劑達8倍量時,含量幾乎達到飽和。再繼續(xù)加大溶劑量,雖收率有所增高,但因浸出了雜質導致最終評分下降。因此,綜合考慮將溶劑量定為8倍量較為合適。見圖3。

圖1 提取時間的影響
2.5 回歸模型的建立和分析 利用Design-Expert 8.0.6對下表數據進行多項式回歸分析,建立枳椇子提取工藝參數的回歸模型,得到總黃酮含量、皂苷含量及收率綜合得分編碼自變量A、B、C的二次多項回歸方程:方程Y=0.014 A+0.56B+0.46C-0.30AB+0.007 5AC-0.49BC+0.038 A2-0.37B2-0.29C2+8.28。對于該模型進行方差分析和模型系數顯著性檢驗,結果見表2。

圖2 提取次數的影響
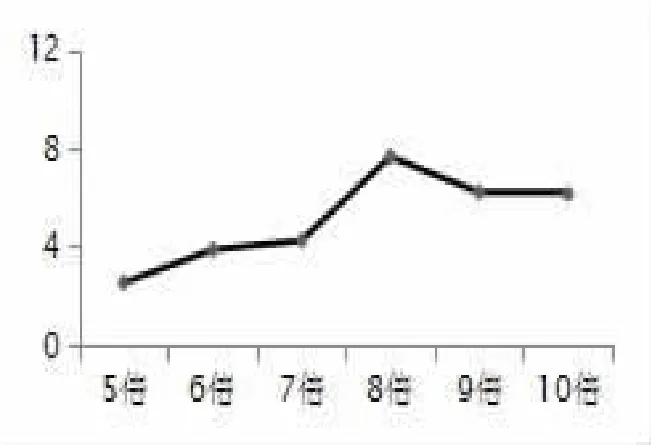
圖3 溶劑倍量的影響

表2 Box-Behnken實驗設計及結果
對該模型進行方差分析及模型系數顯著性檢驗,可見回歸模型極其顯著(P<0.01),說明建立的模型有意義;失擬項P>0.05,表明差異無統(tǒng)計學意義,模型擬合度良好,隨機產生的模型的殘差可以不計,可用此模型和方程來分析預測枳椇子總黃酮含量、皂苷含量及收率的綜合得分。結果見表3。
模型由方差分析結果可知,對回歸方程進行檢驗,模型的矯正系數R2Adj為0.981 9,說明此模型能解釋98.19%響應值的變化,即此數據擬合度很高,試驗誤差小,因此可以用于枳椇子提取工藝的分析和預測。回歸模型系數顯著性檢驗結果中可以看出,模型的一次項中B、C影響顯著,二次項B2影響顯著,其他各項對綜合得分的影響不是簡單的線性關系[19]。
2.6 響應曲面分析與優(yōu)化 響應曲面圖是根據回歸方程繪制的,是響應值在各實驗因素交互作用下得到的結果構成的一個三維空間曲面,可以預測和檢驗變量的響應值以及確定變量的相互關系。AB、AC和BC之間均存在交互作用,根據回歸方程做出模型的響應曲面及等高線見圖4。

表3 回歸模型和方差分析

圖4 等高線和響應面圖
利用Design-Expert 8.0.6軟件求解回歸方程。最高綜合得分為8.787 55,其最優(yōu)提取條件為提取時間為2.00 h,提取次數為4.00次,溶劑倍量為7.86倍,此條件下得到皂苷含量為8.910 1%,總黃酮含量為6.420 7%,干膏收率為11.820 8%,綜合得分8.787 56。
2.7 最佳提取工藝確定及驗證實驗 為了方便,將提取時間定為2小時、提取次數4次、溶劑倍量定為8倍量,依據所確定的最佳提取工藝進行實際驗證(n=3),實際測的枳椇子皂苷平均含量8.879 7%(RSD=1.4%),總黃酮平均含量為6.452 1%(RSD=0.8%),干膏收率為11.117 9%(RSD=0.5%),綜合評分為8.580 12,這與理論預測值(8.787 56)接近;因此,采用Box-Behnken實驗設計優(yōu)化的得到得枳椇子提取工藝條件準確可靠,具有實用價值。
3 結論
本實驗利用響應曲面法,通過二次回歸設計得到枳椇子皂苷含量、總黃酮含量和干膏收率與提取時間、提取次數和溶劑倍量關系的回歸模型。最終得出最優(yōu)提取工藝條件為提取時間2小時、提取次數4次、提取溶劑8倍量,此條件下得到皂苷含量為8.910 1%,總黃酮含量為6.420 7%,干膏收率為11.820 8%。中藥具有多組分、多靶點的特點,通過將多指標綜合評分能夠篩選以多個目標成分在同一提取條件下的提取工藝并進行綜合評價,兼顧各指標的協同效應,減少了單因素分別試驗的局限性,比單一指標篩選更為科學、全面,能夠提高提取效率,減少服用劑量、提高療效要求,更可以保證制劑的質量[20]。
參考文獻
[1]于斌如,湯銀紅.枳椇子的研究進展[J].時珍國醫(yī)國藥,2004,15(9):608-609.
[2]江蘇新醫(yī)學院.中藥大辭典[M].北京:人民出版社,1977:1511.
[3]張洪,劉秀玲,文為.優(yōu)化枳椇子的提取工藝[J].華西藥學雜志,2005,20(12):121-122.
[4]時濤,王曉玲,陳振德,等.枳椇子化學成分及藥理活性研究進展[J].中藥材,2006,29(15):510-513.
[5]Xu WT,Zhang FF,Huang KL,et al.Application of respose surface methodology for extraction optimization of water-soluble polysaccharides form Pteridium aquilinum[J].Food science,2008,29(7):122-126.
[6]于淼,柏云嬌,代岐昌,等.響應曲面法優(yōu)化文殊蘭中生物堿的提取工藝[J].中草藥,2013,44(10):1286-1289.
[7]劉艷杰,項榮武.星點設計效應面法在藥學試驗設計中的應用[J].中國現代應用藥學雜志,2007,24(6):455-457.
[8]Pan Y,Cai BC,Wang KL,et al.Neferine enhances insulin sensitivity in insulin resistant rats[J].J Ethnopharmacol,2009,124(1):98-102.
[9]Guan J,Fang Q,Hanna MA.Selected functional properties of extruded starch acetate and natural fibers foams[J].Cereal chemistry,2004,81(2):199-206.
[10]楊文雄,高彥祥.響應面法及其在食品工業(yè)中的應用[J].中國食品添加劑,2005,25(2):62-71.
[11]劉穎新,劉利利,喻祖文,等.星點設計-響應面法優(yōu)化厚樸提取工藝[J].中國實驗方劑學雜志,2013,19(7):27-30.
[12]蔣紅,王宏軍,李繼昌,等.大葉南五味子中水溶性多糖提取工藝條件優(yōu)化[J].東北農業(yè)大學學報,2012,43(6):60-64.
[13]孔濤,范杰平,胡小芳,等.響應面法優(yōu)化超聲輔助提取車前草中的熊果酸[J].食品科學,2011,32(6):80-84.
[14]胡志林,劉文娜,張永軍.枳椇子中總黃酮提取工藝研究[J].中成藥,2004,26(12):1065-1067.
[15]張紀立,黃儀鳳,程榮珍,等.絞股藍皂苷含量測定方法比較[J].中成藥,1995,17(4):39-40.
[16]王振忠,武文潔.野菊花總黃酮提取工藝的響應面設計優(yōu)化[J].時珍國醫(yī)國藥,2007,18(3):648-650.
[17]張小梅,梁旭明,楊榮平,等.反相高效液相色譜法測定吳茱萸中吳茱萸堿和吳茱萸次堿含量[J].中國藥業(yè),2011,20(9):29-30.
[18]曾金祥,劉勇,黃碧濤,等.吳茱萸HPLC指紋圖譜的建立及3 種成分含量的測定[J].安徽農業(yè)科學,2011,39(14):8273-8278.
[19]王群,劉文,宋信莉,等.多指標優(yōu)化吳茱萸的提取純化工藝[J].中國試驗方劑學雜志,2013,19(3):45-47.
[20]曾金祥,魏娟,張壽文,等.響應面-多指標評價法優(yōu)化吳茱萸提取工藝[J].中國新藥雜志,2014,23(15):1819-1823.
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
山東冶金(2019年6期)2020-01-06 07:45:54
世界農藥(2019年2期)2019-07-13 05:55:12
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
光學精密工程(2016年6期)2016-11-07 09:07:19
銅業(yè)工程(2015年4期)2015-12-29 02:48:39
