微波萃取結合高效液相色譜-電感耦合等離子體串聯質譜同步分析水中砷、硒和鉻形態
2018-07-24 08:45:30林曉娜何衛東張焊宇魏霄凌周曉蓮俞夢翔
食品科學 2018年14期
林曉娜,戴 騏,何衛東,張 芳,張焊宇,魏霄凌,周曉蓮,俞夢翔
(浙江省檢驗檢疫科學技術學院,浙江 杭州 310016)
元素化學形態是環境、毒理學和分析領域的一個重要話題[1]。元素形態分析在總量分析的基礎上得到了附加信息,包括生物利用度、遷移率、代謝過程、生物轉化過程和相關毒性等,通過元素形態分析可以更清晰地理解元素總量的重要性[2-5]。工業廢水污染和紡織印染工業中人造纖維織物等行業的發展導致多種毒性或微弱或劇烈的砷化物及其衍生物、鉻(VI)等通過地質沉積等地球化學過程進入到土壤和飲用水水源[4]。一些不良生產者和商家在所謂的富硒食品、農產品乃至保健品中添加低成本的無機硒(亞硒酸鈉和硒酸鈉)而非真正有機硒。砷形態中無機砷毒性高,有機砷被甲基化后不具有毒性[2]。硒和鉻是人類生存的必需微量元素,不管是過量攝入或者缺乏都會引起嚴重的生物學和生態學問題,例如硒過量攝入會導致硒中毒,缺乏則會引發慢性克山病[4]。硒的毒性取決于它的氧化形態和數量。硒的多種存在形態中,單質硒實際上是無毒的,因為它既不易溶解又不易吸收,亞硒酸毒性最強,硒酸次之,硒代氨基酸反而對人體有益[4]。鉻無法在人體內合成,只能從外源吸收,同硒一樣,鉻的缺乏和過量都可能會引起疾病,甚至死亡[6]。鉻(VI)很容易出現在土壤-水系統中的生物體[7]。因此砷、硒和鉻形態分析比總量分析更為重要,重金屬的危害程度取決于其存在的濃度及化學形態[6]。近年來元素形態分析受到了科研部門和商業實驗室的重視,應用最多的是高效液相色譜-電感耦合等離子體質譜聯用(high performance liquid chromatography-inductively coupled with plasma mass spectrometry,HPLC-ICP-MS)和氣相色譜-電感耦合等離子體質譜聯用(gas chromatography inductively coupled with plasma mass spectrometry,GC-ICP-MS),對鉻(VI)、無機汞、甲基汞、無機砷和有機錫檢測均已被美國環保署方法認同。
生活飲用水是人類生活的重要部分,容易受到環境土壤的直接影響。根據中國的飲用水質量標準,砷、鉻(VI)和硒的限量分別為0.01、0.05 mg/L和0.01 mg/L,歐盟飲用水條例(98/83/EC)也有相同的要求。
本研究旨在建立一種同時分離和測定生活飲用水中的砷、硒和鉻元素形態的方法,為食品中砷、硒和鉻的形態分析提供基礎研究。有研究報道HPLC-ICP-MS同時測定砷,硒和鉻無機形態,但并不是使用單一前處理方法和同時測定的[8]。目前,在眾多的聯用技術中因HPLCICP-MS具有高效的分離能力和高靈敏度的檢測能力,成為元素形態分析中應用最廣泛的技術。本實驗使用的電感耦合等離子體串聯質譜(inductively coupled plasma tandem mass spectrometry,ICP-MS/MS)技術[9-10],其性能優于單四極桿ICP-MS,具有兩個獨立質量篩選功能的過濾器,可控且持續的干擾消除能力,不僅具備ORS3氦碰撞動能歧視消干擾的性能,其MS/MS功能還能精確控制進入碰撞/反應池的離子,有效解決了原先傳統反應池在使用反應性氣體測定復雜基體時因共存基體或元素易形成新的干擾離子或共存離子導致用質量轉移法難以獲得準確的痕量定量結果等難點,可以有效解決復雜基質中的超痕量元素所受的質譜干擾難題,即使樣品組成有所不同,也能保持一致、可預見的反應條件,能始終有效地消除復雜樣品中未知元素對超痕量目標元素的干擾[11-13]。
1 材料與方法
1.1 材料與試劑
超純水(18.2 MΩ)制備包括流動相的所有標準品和溶液。
標準物質:砷甜菜堿(arsenic betaine,ASB)(GBW08670,0.518 μmol/g)、砷酸根(arsenate,As(V)GBW08667,0.233 μmol/g)、亞砷酸根(As(III),GBW08666,1.011 μmol/g)、一甲基砷(methyl arsenic,MMA)(GBW08668,0.335 μmol/g)、二甲基砷(dimethyl arsenic,DMA)(GBW08669,0.706 μmol/g)、硒酸根(Se(IV),GBW10033,0.525 μmol/g)、亞硒酸根(Se(VI),GBW10032,0.543 μmol/g)、三價鉻(Cr(III),GSB04-1723-2004(e),1 000 μg/mL)、六價鉻(Cr(VI),GBW(E)080257,100 μg/mL)。以上標準溶液均購自國家標準物質中心。
0.5 mmol/L EDTA緩沖溶液,用20%氨水調節pH值至7.5。在分析前約1 h用緩沖溶液制備含有混合砷、硒和鉻形態標準工作溶液,室溫條件下至少反應45 min,最大效率地形成[CrEDTA]-絡合物。
1.2 儀器與設備
1260 Infinity HPLC儀、8800 ICP-MS/MS儀美國Agilent Technologies公司;PRP-X100陰離子交換柱(250 mm×4.1 mm,10 μm) 美國Hamilton公司。
1.3 方法
1.3.1 HPLC條件
形態分離采用HPLC儀,在樣品流路中采用了無金屬部件及溶劑輸送無鐵、無鋼的設計,最大程度上減少了鉻形態分析時引入污染。Hamilton PRP-X100陰離子交換柱(250 mm×4.1 mm,10 μm)進行分離。HPLC系統的梯度程序見表1。
1.3.2 ICP-MS/MS條件
應用8800 ICP-MS/MS儀,采用1.0 μg/L調諧液優化氦模式(He)和高能He(high energy He,HEHe)模式儀器參數,包括氧化物、雙電荷和靈敏度;5.0 μg/L砷、硒和鉻標液優化加氧模式參數,包括靈敏度及反應氣體流量[14]。儀器的操作條件及參數見表2、3。

表2 HPLC-ICP-MS的儀器條件Table2 Instrumental conditions for HPLC-ICP-MS
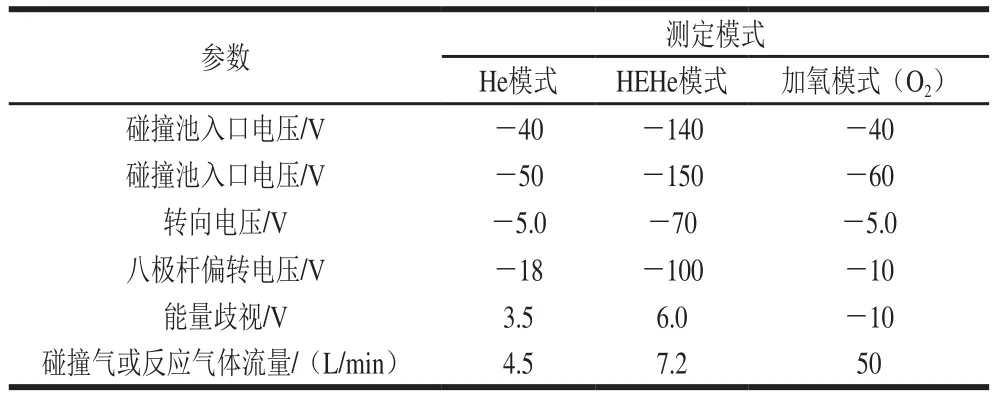
表3 ICP-MS/MS不同測定模式下的特殊儀器參數Table3 Special instrumental parameters for different measurement modes of ICP-MS/MS
2 結果與分析
ICP離子源中形成的同質異位素、雙電荷離子、氬離子、多原子離子都會對相同質量數的離子測量產生影響,質量數小于80的元素受到干擾尤為明顯,75As+、78Se+和52Cr+的質譜干擾有40Ar35Cl+、78Gd++、40Ar38Cl+和40Ar12C+等。降低等離子體功率、提高儀器的分辨率可以消除部分干擾,但其代價是損失靈敏度;碰撞、反應氣的引入,也可能形成新的干擾影響因素。本研究使用串聯質譜測定砷、硒和鉻,在反應池加氧之前先經過第一級四極桿(Q1)進行初篩,在Q1后的碰撞反應池(八極桿)加入氧氣,75As+、78Se+和52Cr+與16O反應生成91AsO+、94SeO+和78CrO+,由于Q1的特異篩選作用,m/z 91、94和78的離子和多原子離子(94Zr+、59Co35Cl+)已在Q1時被過濾,未能進入Q2,從而達到徹底消除同質異位素、雙電荷離子、氬離子、多原子離子的干擾[15-17]。
2.1 反應氣流量優化及消除干擾效果
通過考察在加氧條件下各質量數的背景等效濃度(background equivalent concentration,BEC),使元素的氧化效率最大化,且有效降低背景,同時保證足夠的靈敏度(檢出限),以5 μg/L的As(75→91)進行反應氣體流量優化結果見圖1,隨著氧氣流量增加,As的氧化效率(靈敏度)和BEC均出現先提高后下降的趨勢:氧氣流量達到45%時氧化效率最大,75As(40 517.73 cps)比He模式下75As(6 225.54 cps)提高6.5 倍,隨著反應池中氧氣繼續增加,產生了空間電荷效應,砷的氧化效率受到抑制;25%的氧氣流量時,As(75→91)的BEC最低(0.0128 μg/L),隨著氧氣流量增加,背景離子的氧化效率提高,BEC提高。Se和Cr的反應氣體流量優化結果分別為25%和30%,綜合考慮3 種元素的情況,最終確定砷、硒和鉻的最佳氧氣流量為25%。

圖1 反應氣(氧氣)流量優化結果Fig.1 Optimization of reaction gas (oxygen) fl ow
本研究采用的8800 ICP-MS/MS儀有多種測定模式,包括傳統ICP-MS的No Gas模式、以碰撞和動能歧視降低干擾的氦模式(普通He模式和HEHe模式)、以及加氧氣為反應氣的單桿模式(Q1作為離子導桿,不起到質量篩選作用)和串接模式(使用Q1作為質量過濾器,讓特定質量數離子通過)。為研究串接加氧模式在消除干擾的優勢,本實驗同時研究了砷、硒和鉻在4 種測定模式下的儀器檢出限和BEC,因砷、硒和鉻受載氣和基質形成的多原子離子干擾較嚴重,故不再研究No Gas模式的測定效果。

表4 砷、硒和鉻不同ICP-MS/MS測定模式下的檢出限和BECTable4 Detection limits and background equivalent concentrations of As, Se and Cr in different assay modes of ICP-MS/MS
表4顯示:1)砷、硒和鉻在加氧串接模式下的BEC和檢出限比其他模式低,說明加氧串接模式靈敏度高,消除干擾的能力最強;2)HEHe模式下BEC比串接模式低,這是因為HEHe模式對該同位素也有較強的干擾消除能力,但由于HEHe模式與He模式都是通過He碰撞實現干擾消除,因此靈敏度相對較差;3)單桿氧模式結果說明引入了其他的質譜干擾,證明了Q1具有質量過濾器作用,對消除質譜干擾和提高靈敏度有很大的作用。因此加氧串接模式能徹底地消除質譜干擾,同時還提高了靈敏度[18]。
2.2 色譜分離條件
常規H P L C使用不銹鋼材質,鉻質量分數為12%~30%。流動相淋洗時,難免會有微量鉻被洗脫,而Bio-HPLC凡是和樣品接觸的部件均是由鈦、陶瓷和聚醚醚酮制成,完全避免了HPLC本身引入的鉻污染,因此Bio-HPLC的背景比常規HPLC低一個數量級[19]。
2.2.1 色譜柱選擇
色譜分離條件主要包括色譜柱選擇和流動相。元素的多數形態均為離子形態,常采用離子交換柱、反相色譜配合離子對試劑進行分離。離子對試劑最大的缺點是對色譜柱不可逆的傷害較大。在生物和環境試樣中,元素通常以中性或帶電荷的化合物形式存在,這些形態隨分析溶液的pH值變化而變化,因此離子交換色譜柱應該是最適宜分離柱。通過文獻查閱以及試驗確定本研究選用Hamilton PRP-X100陰離子交換色譜柱,其他色譜柱留待進一步研究。
2.2.2 流動相的組成
流動相是影響HPLC分離的另一個主要因素,主要優化參數有pH值、有機相、離子對試劑和梯度。流動相的組成選擇原則:提供洗脫分析物所需的離子強度,不干擾目標分離物,并且不會導致ICP離子化效率的降低,盡量避免使用鈉鹽、鉀鹽等含常量元素。實驗發現NH4H2PO4、NH4HCO3和NH4NO3等流動相均能實現砷形態分離,且碳酸鹽的分離時間較磷酸鹽短,但NH4H2PO4對Se和Cr形態均不洗脫。NH4NO3常用于Cr(III)和Cr(VI)的分離,但NH4NO3是氧化劑,對無機砷和無機硒有氧化作用,初步實驗未找到合適的濃度和pH值可用來同時分離砷、硒和鉻形態。而NH4HCO3可實現As(III)、As(V)、Se(IV)、 Se(VI)、Cr(III)和Cr(VI)的分離。
在流動相中加入甲醇等有機物能夠改進As和Se的靈敏度,但是會在等離子體中形成40Ar13C導致Cr信噪比的顯著降低,影響鉻的檢出限,并且碳積聚還會堵塞ICPMS錐口,增加儀器系統維護工作頻率[20]。由于氧氣反應氣的加入,As和Se的靈敏度已大大提高,因此本實驗流動相不再考慮使用有機物。
流動相濃度對分析物的分離時間有影響:實驗通過改變NH4HCO3濃度從0.1~10 mmol/L進行等度洗脫,用氨水調節流動相pH 7.5。NH4HCO3濃度低,As(V)、Se(VI)和Cr(VI)的保留時間較長(24 min),濃度提高后As(III)和DMA保留時間縮短,但分離度不佳。使用等度洗脫,砷形態實現良好的基線分離以及短的分析時間是不可行的。通過實驗確定使用Hamilton PRP-X100,流動相A為4 mmol/L NH4HCO3溶液和流動相B為0.3 mol/L NH4HCO3溶液梯度程序(表1)的HPLC條件下,可實現多種砷、硒和鉻形態的分離。
2.2.3 流動相pH值
基于文獻研究,Cr(VI)在堿性條件下因氧化能力降低而更加穩定[12]。水中存在一定量的氯化物,因此在標液中加入1 000 mg/L氯化物作為干擾物研究流動相pH值(研究范圍7.0~10.2)對砷、硒和鉻形態分離的影響。氯化物形成的干擾物分別為40Ar35Cl+、36Cl16O+和40Ar37Cl+。如圖2所示,隨著pH值的增加,干擾物保留時間不變,但是砷、硒和鉻形態的保留時間有變化。pH 7.5時,Se(IV)與干擾物部分重疊;pH 8.2時MMA受干擾物影響;pH值提高至8.7時MMA與干擾物實現分離;pH 9.2時As(III)作為陰離子化合物存在(pKa9.2),并被固定相保留,隨著pH值升高至10.2時,As(III)與MMA完全重合。pH值的變化。氯化物干擾物對鉻形態均無干擾。在0~3 min時降低流動相A的濃度,可以實現As(III)與MMA的基線分離。但隨著pH值升高,Cr(III)和Cr(VI)的分離變差。
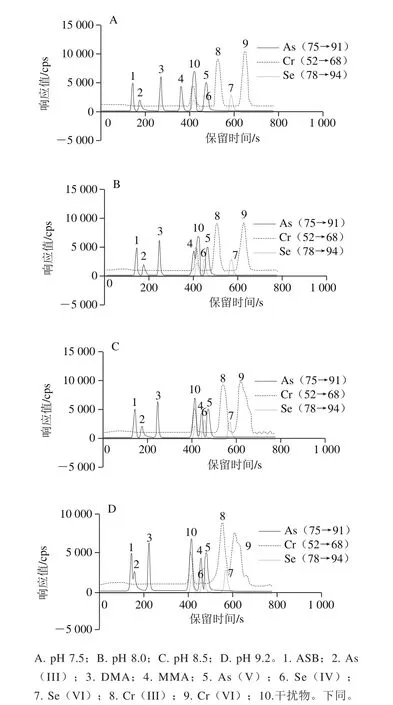
圖2 不同pH值下砷、硒和鉻形態的分離效果Fig.2 Separation of arsenic, selenium and chromium at different pH values
pH值對砷、硒和鉻形態分離非常重要[15]。采用單四極桿ICP MS在克服質譜干擾的情況下,只在pH 8.5時可以同時分離As(III)、DMA、MMA、As(V)、Se(IV)、Se(VI)和Cr(VI)7 種形態,但Cr(VI)和 Cr(III)的峰形和分離效果不佳,需要考慮其他的方式來克服質譜干擾。串聯質譜因其高效特異準確的質譜干擾消除能力,可解決這一難題。pH 7.5,采用串聯質譜進行測定(圖3),結果發現氯化物干擾物未出現在色譜圖上,ASB、As(III)、DMA、MMA、As(V)、Se(IV)、Se(VI)、Cr(III)和Cr(VI)實現基線分離,從譜圖還能看到砷和硒的靈敏度提高,鉻的BEC也有所下降。

圖3 HPLC-ICP-MS/MS分離砷、硒和鉻形態效果圖Fig.3 Separation of different species of arsenic, selenium and chromium by HPLC-ICP-MS/MS
經過質譜條件和色譜條件優化后確認采用生物惰性HPLC,以Hamilton PRP-X100為色譜柱,以4 mmol/L NH4HCO3(A)和0.3 mol/L NH4HCO3(B)為流動相(pH 7.5),梯度洗脫分離砷、硒和鉻的9 種元素形態后,ICP-MS/MS進行測定,有效地消除了質譜干擾并提高了靈敏度。
2.3 樣品檢測結果
適宜的提取方法是形態分析的關鍵環節,樣品基質不同最優的提取方法不同,砷形態按照提取液的不同,可分為水提取、酶提取、甲醇-水提取、Tris-HCl提取、氯仿-甲醇-水提取、稀硝酸提取[21-23];硒形態的提取,目前有酶水解法、加壓液相提取法、HNO3-H2O2微波提取法、水-胃蛋白酶-胰蛋白酶三步提取法等[24-25]。鉻形態的提取,有非氧化性無機酸(如HCl、H3PO4)、還原性更弱的有機酸(如乙酸、甲酸等)、堿液(如四丁基氫氧化銨、氨水)、EDTA等[26-28]。
砷和硒的幾個形態均以陰離子形式存在,鉻元素主要以Cr(III)和Cr(VI)的形態存在。其中Cr(III)是陽離子,Cr(VI)是鉻酸根陰離子[29]。在相同條件下,采用一種離子交換方法是不能對這兩種離子都起作用的[30]。EDTA是Cr(III)的有效絡合劑,前處理時加入EDTA緩沖液能與Cr(III)形成穩定的 Cr(III)-EDTA絡合物,Cr(VI)不與EDTA 絡合,直接溶于緩沖液中。Cr(III)和Cr(VI)就分別以Cr(III)-EDTA和Cr(VI)形態在色譜柱中分離。
EDTA是一種常用的絡合劑,它既能與Cr(III)絡合,也能與鈣、鎂、鋅、鐵等多種金屬離子進行絡合,它們之間是一種競爭關系。因此,如果EDTA加入量不夠,就會導致Cr(III)的回收率偏低。EDTA與金屬離子通常以1∶1絡合的。通過實驗發現Cr(III)的濃度趨勢隨著EDTA濃度的增加而增加(圖4),EDTA濃度0.5 m o l/L后趨于穩定,因此E D T A濃度確定用0.5 mol/L,同時要用氨水調節水溶液pH 7.5,避免Cr(VI)被還原為Cr(III)。

圖4 EDTA濃度對Cr(III)絡合影響Fig.4 Effect of EDTA concentration on Cr(III) complexation
Cr(III)與EDTA在常溫下絡合速度非常緩慢,需要幾十小時才能完全絡合。隨著溫度升高,絡合速度明顯加快。在水中加入10 μg/L的形態混合標液,前處理方法見表5。考察恒溫振蕩器、微波萃取和超聲波提取效果,結果見表6。砷和硒各種前處理方法回收率都較好,這可能是水中砷、硒含量較低,溫度提高,Cr(III)的絡合時間短。100 ℃條件下能極大地加快絡合速度,時間越長,Cr(VI)部分轉化為Cr(III),導致Cr(VI)回收率下降。因此,100 ℃微波萃取2 min后Cr(III)能與EDTA完全絡合。

表5 水的前處理方法Table5 Pretreatments for water samples

表6 不同制備方法的砷、硒和鉻回收率結果Table6 Recoveries of arsenic, selenium and chromium with different preparation methods
2.4 檢出限、精密度和回收率實驗結果
9 種元素形態在0~50 μg/L范圍內線性良好,線性相關系數大于0.999。根據峰面積信噪比實驗確定檢出限分別為0.023(AB)、0.056(As(III))、0.032(DMA)、0.064(MMA)、0.061(As(V)),0.091(Se(IV))、0.089(Se(VI))、0.21(Cr(III))、0.18(Cr(VI))μg/L。各元素檢出限遠低于中國飲用水水質標準限量。通過3檔加標5 次平行的回收率(72%~119%)和精密度(0.23%~11.9%)實驗,結果均符合要求,見表7。

表7 砷、硒和鉻形態的回收率和精密度結果(n=5)Table7 Recovery and precision of arsenic, selenium and chromium species from spiked water samples (n= 5)
應用本方法測定了來自杭州不同區域的10 個地表水,情況如下:所有水中均未檢出MMA和ASB,有4 個水檢出DMA,7 個水檢出AsV,9 個水檢出As(III),檢出的砷形態濃度大致在0.1~2.0 μg/L;所有的硒均未檢出;有1 個郊區的水檢出了8 μg/L的Cr(VI),經調查,可能是該區域之前有個化工廠導致的污染。
3 結 論
本研究采用微波萃取結合HPLC-ICP-MS/MS法測定生活飲用水中的砷、硒和鉻形態,針對飲用水中的As(III)、As(V)、ASB、DMA、MMA、Se(IV)、Se(VI)、Cr(III)和Cr(VI)進行了分離檢測。使用NH4HCO3進行梯度洗脫,在13 min內完成分離檢測。本方法的建立為食品中砷、硒和鉻的同步分離提供了理論支持。
