高效液相色譜法測定飼料添加劑D-生物素的含量
2018-11-09 05:40:06裘丞軍任玉琴
中國飼料 2018年19期
關鍵詞:方法
裘丞軍,任玉琴
(浙江省獸藥飼料監察所,浙江杭州311199)
D-生物素即生物素,又稱維生素H,輔酶R,屬于水溶性B族維生素,廣泛存在于動植物組織內。其主要作為體內多種羧化酶的輔酶,參與二氧化碳的轉移固定和羧化反應,促進糖類、脂肪和蛋白質代謝。D-生物素已被收錄于中華人民共和國農業部公告 第2045號《飼料添加劑品種目錄(2013)》,大量應用于生物醫藥、食品化妝中,在畜禽養殖應用中有促進動物正常生長,提高飼料轉化率等重要作用(陳宏等,2008)。目前食品和飼料中生物素含量的測定方法,有微生物測定法(國家標準,GB 5413.19-2010)、熒光免疫層析法(劉海英,2016)、酶聯免疫法(徐幼平等,2000)、分光光度法 (國家標準,GB/T 17778-2005)、高效液相色譜法(潘一斌等,2009;余林梁等,2003)、高效液相色譜-串聯質譜法 (劉進璽等,2010)。其中微生物法、熒光免疫層析法和酶聯免疫法操作繁瑣,試劑昂貴,對檢測技術要求比較高,主要是針對檢測D-生物素含量較低的樣本;分光光度法操作步驟多,時間長;液質聯用法對儀器的要求比較高;而液相色譜法適用范圍大,應用比較廣泛。目前,D-生物素原料的分析方法主要有酸堿滴定法(歐洲藥典8.0版,2014:1671-1672; 日本藥典 16 版,2011:458-459)、高效液相色譜法 (美國藥典 36版,2013:2664-2665),國內飼料行業尚無國家標準。本研究為進一步優化相關條件建立高效液相色譜法,測定不同來源和批次的生物素原料含量,并與酸堿滴定法進行了對比研究,結果確定該方法簡便、快速、靈敏、準確、專屬性好,可以作為生物素飼料原料的質量控制方法。
1 材料與方法
1.1 試劑和儀器 生物素標準品 (中國食品藥品檢定研究院);一水合高氯酸鈉(分析純);磷酸(分析純);三氟乙酸(色譜純);乙腈(色譜純);水(超純水);酚酞指示劑;0.1 mol/L NaOH標準滴定液。
飼料添加劑D-生物素樣品由四家企業提供( 批 號 :141102、VH20150328、CS1-1502019、KXVHUSP150901)。
Agilent1260高效液相色譜儀,紫外檢測器(美國Agilent公司);KQ-500E超聲儀(昆山超聲儀器公司);ZP205電子天平(Mettler公司)。
1.2 色譜條件 色譜柱:Agilent XDB-C18柱,柱長 150 mm,內徑 4.6 mm,粒徑 5μm;流動相:乙腈+緩沖液(含0.1%高氯酸鈉和0.1%磷酸的水溶液)=8.5+91.5(體積比)。 流速:1.2 mL/min;進樣量:20 μL;紫外檢測波長:210 nm。
1.3 標準溶液配制 準確稱取生物素標準品40 mg(精確至0.01 mg),置于200 mL容量瓶中,加入提取液(乙腈+水=1+4)約 150 mL,50℃水浴超聲波助溶5 min,冷卻至室溫,用提取液定容至刻度,混勻。
1.4 飼料添加劑樣品前處理 準確稱取飼料添加劑試樣 40 mg(精確至 0.01 mg),置于 200 mL容量瓶中,加入提取液(乙腈+水=1+4)約150 mL,50℃水浴超聲波助溶5 min,冷卻至室溫,用提取液定容至刻度,混勻。
2 結果與分析
2.1 方法線性和檢測限試驗 取適量標準品溶解稀釋成 25、50、100、200、400 μg/mL 系列濃度的溶液,取20μL注入液相色譜測定分析,記錄生物素峰面積,以濃度(x)為橫坐標,峰面積(y)為縱坐標,繪制線性回歸曲線。結果表明生物素在25~400μg/mL質量濃度范圍內線性關系良好,y=7.46044x+4.06379(r=1.00000),方法檢測限為1.5 mg/kg,滿足檢測要求。標準品色譜圖見圖1。
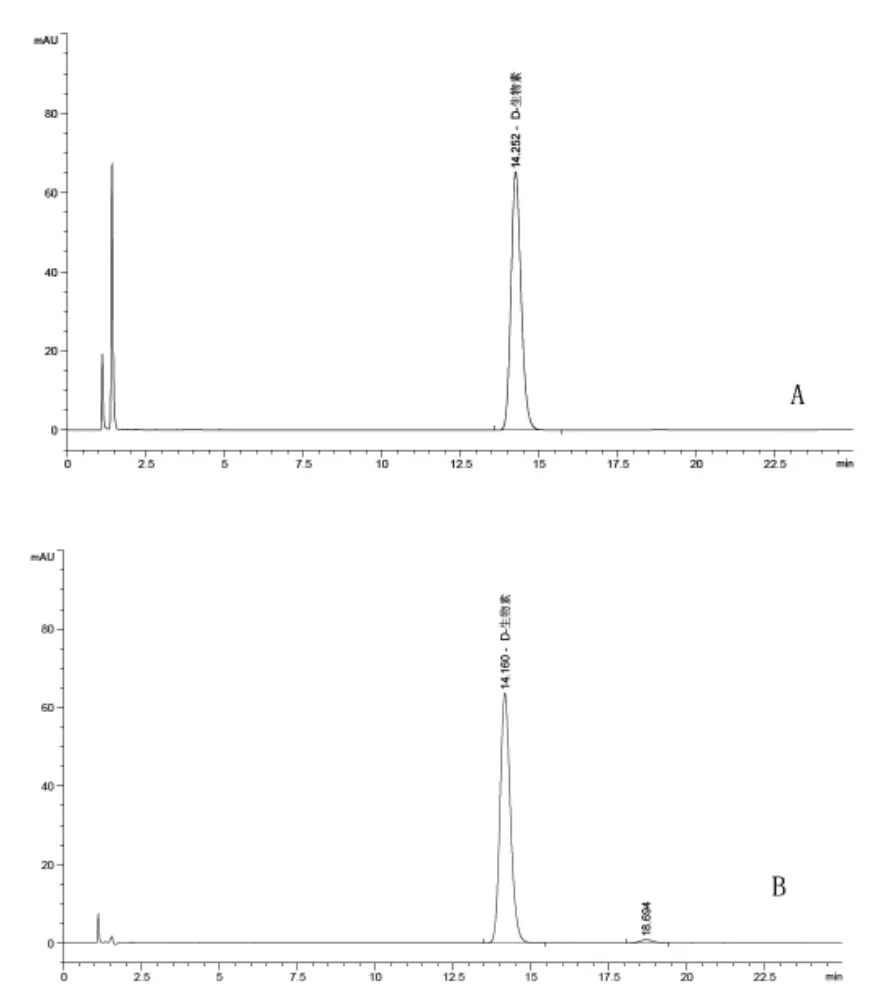
2.2 精密度試驗 取四批次樣品 (批號141102、VH20150328、CS1-1502019、KXVHUSP150901)各5份,按試驗方法1.4處理,取20μL樣品溶液注入液相色譜測定分析,外標法計算含量,結果如表2, 平均含量分別為 100.4%、99.3%、99.9%、95.9%,RSD(n=5)為 0.1%、0.2%、0.1%、0.2%,表明精密度良好。
2.3 準確度試驗 取生物素標準品約30、40、50 mg各3份(精確至0.01 mg),按試驗方法1.4處理,取20μL樣品溶液注入液相色譜測定分析,外標法計算含量,并計算回收率,結果見表1。
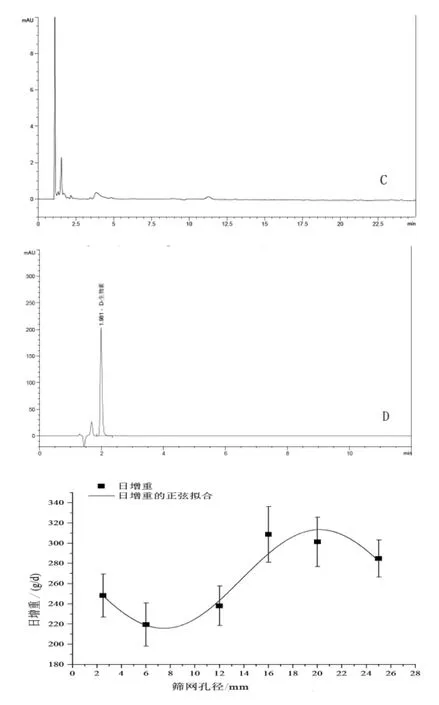
圖1 HPLC色譜圖
2.4 與酸堿滴定法測定含量的比較 參照美國藥典36版 (2013:2664-2665) 測定生物素的方法,稱取四批次樣品(批號CS1-1502019、141102、VH20150328、KXVHUSP150901)各 5 份,每份約500 mg(精確到0.1 mg),置于 250 mL錐形瓶中,加水100 mL。加熱攪拌溶解,加酚酞指示劑2滴,用0.1 mol/L NaOH標準滴定液緩慢滴定至粉紅色,且保持30秒不褪色,每毫升0.1 mol/L NaOH標準滴定液相當于24.43 mg的生物素。計算樣品含量和RSD,結果如表2,試樣1與試樣2兩種方法含量測定結果基本一致,試樣3和試樣4用本方法測定的結果較酸堿滴定法低。進一步分析試樣溶液色譜圖,發現試樣1和2色譜圖基線干凈無雜峰,而試樣3和4主峰后有雜質峰出現,并呈完全的基線分離,表明酸堿滴定法定量沒有專屬性,準確度較低,而本方法分離效果好,準確度高。另外RSD數據表明本方法的精密度比酸堿滴定法相對更高,所以確定本方法定量更加準確合理。
表1 回收率試驗結果(n=9)
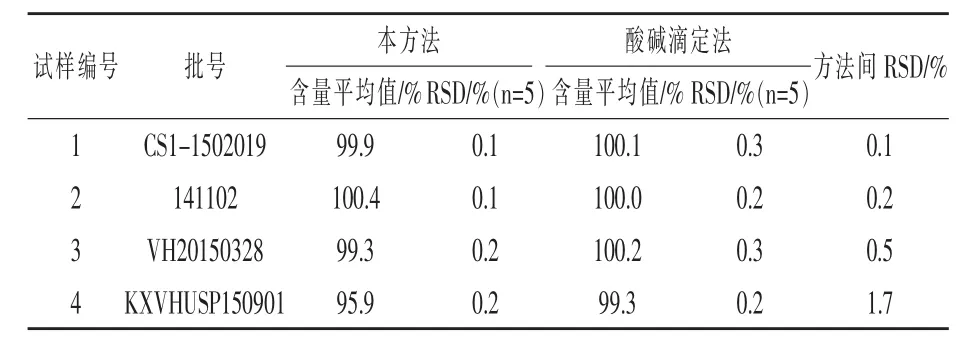
表2 兩種含量測定方法結果比較
2.5 溶液放置穩定性 取樣品 (批號KXVHUSP150901)按試驗方法1.4處理,在室溫分別放置 0、25、50、100、150、250 min 后測定,記錄峰面積,顯示峰面積分別為1472、1469、1472、1470、1473、1472 (單位 mAU*s);RSD=0.1%(n=6)。結果表明樣品溶液在考察的時間范圍內有很好的穩定性。
2.6 專屬性 取樣品(批號CS1-1502019)約20 mg(共5份),其中三份分別加5 mL 1 mol/L鹽酸溶液、5 mL 1 mol/L氫氧化鈉溶液、5 mL 30%雙氧水,放置18 h,進行酸破壞、堿破壞、氧化破壞,pH調至中性后用提取液稀釋至100 mL;另兩份直接用提取劑稀釋后,一份在254、365 nm紫外波長光照24 h,一份不做任何處理作為對照。結果表明樣品對酸、堿和光照均較穩定,對于氧化破壞較明顯,產生的氧化破壞產物在該色譜條件下完全分離無干擾。色譜圖見圖2。
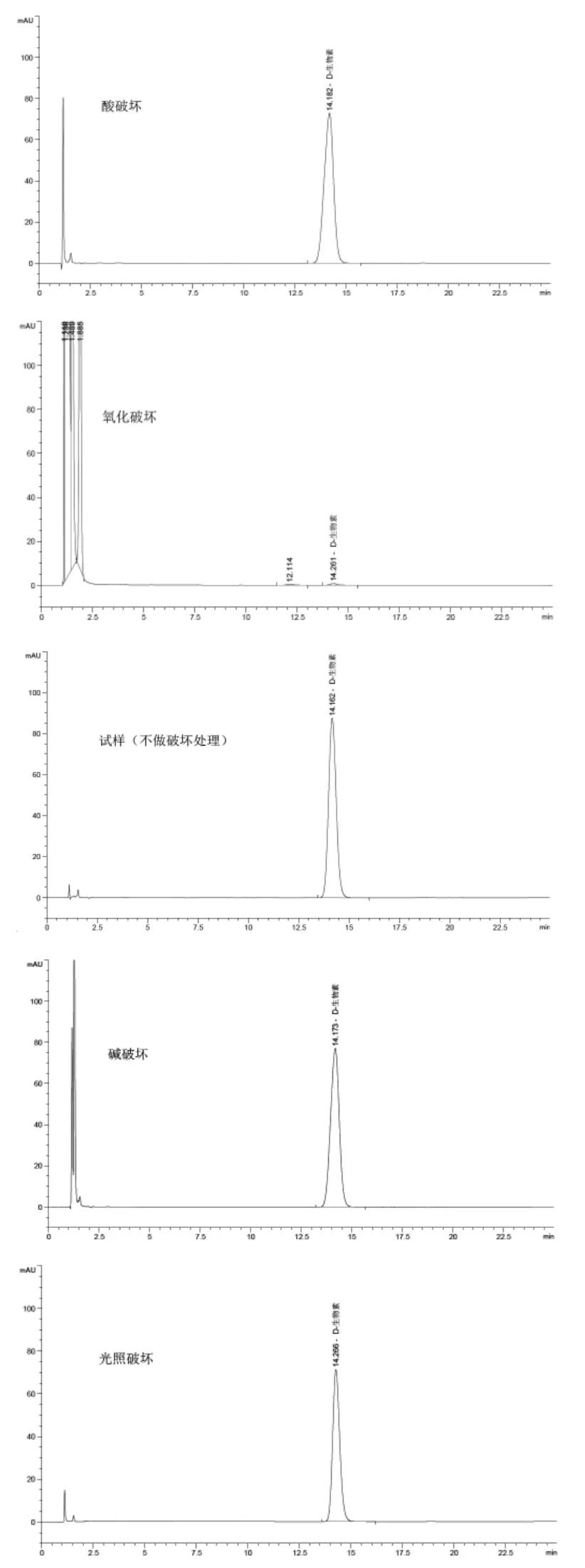
圖2 試樣破壞試驗處理后色譜圖
2.7 耐用性 取樣品(批號KXVHUSP150901)按試驗方法處理,在該色譜條件下通過改變色譜柱型號規格、流動相比例以及色譜儀器型號考察,結果表明色譜系統耐用性良好。
3 討論
3.1 色譜條件優化
3.1.1 波長選擇 生物素在紫外波長光區內沒有特征吸收峰波長,在190~235 nm波長處吸光度由高到低。試驗中選取乙腈/0.05%三氟乙酸溶液=15/85、乙腈/水=1/4、乙腈/0.1%高氯酸鈉和 0.1%磷酸溶液=8.5/91.5三種溶媒,取4批次試樣,每批試樣3份用相應的溶媒溶解稀釋成濃度約為40μg/mL的溶液,用200、210 nm兩個檢測波長,進行5次重復檢測,結果見表3。在乙腈/水作為溶媒時,210 nm吸光度更穩定。因此檢測波長確定為210 nm。
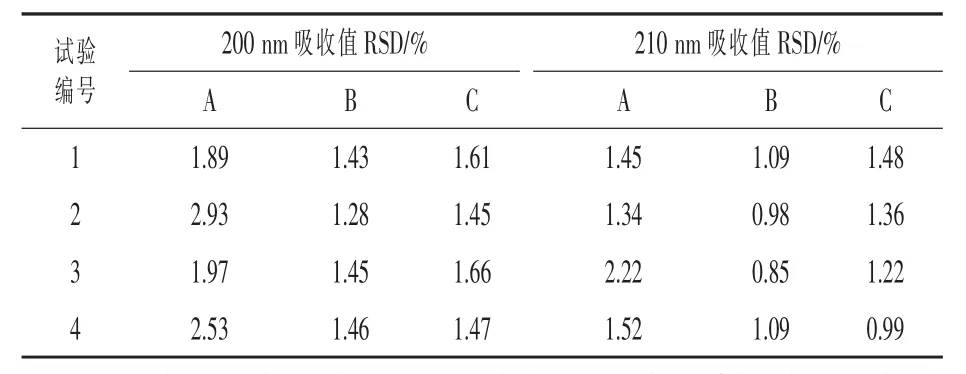
表3 兩種波長和三種溶媒吸光度穩定性比較(n=5)
3.1.2 流動相組成 生物素的化學結構是咪唑酮環與噻吩環相結合的駢環,帶有一個戊酸側鏈,呈弱酸性。在中性和堿性溶液環境中部分或全部以離子形態存在,有較強極性,在C18色譜柱上幾乎不保留,但是在弱酸環境中生物素以分子形態存在,極性減弱,在C18色譜柱上保留時間長。因此選擇帶弱酸性的溶液作為流動相,并且選用在短波長處吸收小的乙腈體系作為流動相。試驗參考美國藥典 36版(2013:2664-2665)選取乙腈磷酸高氯酸鈉緩沖液體系 (乙腈/0.1%高氯酸鈉和0.1%磷酸溶液=8.5/91.5)、參考歐洲藥典8.0版(2014:1671-1672)選取乙腈三氟乙酸溶液體系(乙腈/0.05%三氟乙酸溶液=15/85),用25%乙腈水溶解稀釋試樣至200μg/mL的溶液測試對比分析,并對試劑空白進行色譜分析,結果見表4。生物素在乙腈磷酸高氯酸鈉緩沖液體系中理論塔板數高,對稱因子大,分離度高,保留時間合理,基線噪音小(見圖1),更適合作為流動相。
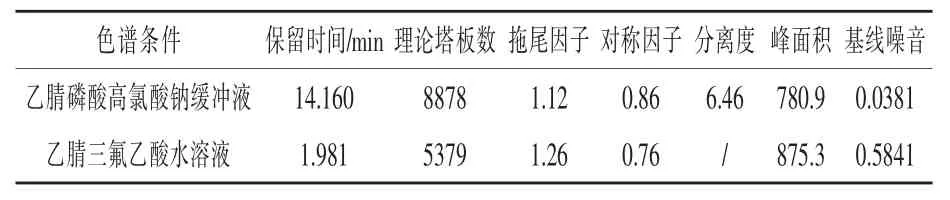
表4 兩種流動相體系比較
3.2 溶解方法的確定 生物素在稀堿中溶解,在水中極微溶,在50℃水中微溶,在乙腈中幾乎不溶,而本方法色譜條件是弱酸性環境,不能用稀堿溶解。結合3.1.1波長選擇試驗,生物素用乙腈水溶液作為溶媒時,吸光度更穩定,因此試驗中精密稱取5份試樣(批號KXVHUSP150901),選取用乙腈/水=1/4作為提取劑,分別直接振搖5 min或者50℃水浴超聲助溶 5、10、20、30 min進行考察分析,測得樣品濃度,結果表明水浴超聲5 min就能夠有效溶解試樣。
4 結論
本文對高效液相色譜法測定飼料添加劑生物素原料的含量進行優化研究,并進一步和酸堿滴定法進行比較,結果表明,酸堿滴定法需人為判斷滴定終點,存在個體差異,專屬性也不強,精密度和準確度相對較差;而本方法選用較為穩定的210 nm為檢測波長,乙腈高氯酸鈉磷酸緩沖液作為流動相,保留時間合理,基線穩定,噪音小,操作簡單,樣品直接溶解后使用高效液相色譜儀外標法定量,人為誤差減少,并且雜質分離效果好,專屬性強,相對酸堿滴定法有較好的精密度和準確度,非常適用于生物素原料的質量控制。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
