甘藍型油菜ALS基因啟動子克隆及其瞬時表達分析
2019-01-09 06:51:08熊冬琴黃子棟張潔夫浦惠明
江蘇農業科學 2018年23期
熊冬琴, 黃子棟, 彭 琦, 張 維, 張潔夫,3, 浦惠明,3, 陳 松,3
(1.江蘇省農業科學院經濟作物研究所,江蘇南京 210014; 2.農業部長江下游棉花與油菜重點實驗室,江蘇南京 210014;3.江蘇省現代作物生產協同創新中心,江蘇南京 210014;4.江蘇農林職業技術學院,江蘇句容 212400)
油菜是世界上最主要的油料作物之一,也是我國重要的油料作物。我國油菜面積、總產量約占世界的1/3,是最大的油菜生產國。油菜油約占我國食用油市場的50%,是我國最主要的食用植物油。鑒于油菜在油料作物中的重要地位,油菜科學的研究一直受到學者們的高度重視。過去幾十年來,油菜遺傳改良已經取得顯著成果,高產、優質雙低油菜品種在生產上已經占據主導地位。近幾年來,隨著現代生物技術與分子生物學研究的飛速發展,一些生物技術在油菜基礎與應用研究中也得到了廣泛應用,如在油菜的分子標記輔助育種、高密度遺傳圖譜構建、重要性狀基因的遺傳定位與克隆,以及基因工程改良等研究領域都取得了顯著進展。
有關油菜啟動子的研究近幾年來不斷有報道。景寅利用反向PCR方法擴增獲得肌醇半乳糖苷合成酶基因(BnGOLS1)的啟動子,能驅動β-葡萄糖苷酸酶(GUS)基因在油菜組織中表達[1];朱斌等用PCR擴增方法獲得甘藍型油菜MAPK7基因家族的啟動子[2];王軒鵬等根據基因組序列獲得甘藍型油菜RabGDI3基因的啟動子,該啟動子能驅動報告基因gus僅在花藥中特異性表達,是組織特異性啟動子[3];陽永學等通過PCR克隆獲得油菜半胱氨酸蛋白酶家族基因BnCP51的啟動子序列,通過轉基因功能驗證該啟動子僅在花蕾、花藥器官中表達,也是一種器官特異性表達啟動子[4];邵鐵梅等基于油菜基因組信息,利用PCR方法克隆到油菜油體蛋白基因的啟動子,該啟動子具有種子特異性表達特性,這也是繼油菜Napin啟動子后的又一個油菜種子特異性表達的啟動子[5];肖旦望等用PCR方法擴增獲得油菜溶血磷脂酰轉移酶基因(LPAT)啟動子[6]。隨著相關研究的進一步開展,會有越來越多的油菜啟動子被發現與應用。
植物中乙酰乳酸合成酶(ALS)是催化分支氨基酸如纈氨酸、亮氨酸和異亮氨酸生物合成第一步的酶。研究發現,抑制植物細胞內的ALS活性,會阻礙支鏈氨基酸(纈氨酸、亮氨酸和異亮氨酸)的生物合成,從而抑制植物細胞的分裂和生長,嚴重的會導致植物死亡。基于這個原理,一些化學公司陸續開發出了一系列ALS抑制劑類的除草劑并在生產上廣泛應用。此外,ALS基因也受到生物學家的普遍關注,一些植物或作物的ALS基因被克隆[7-8]。研究發現,ALS基因編碼序列發生堿基突變,則產生對ALS抑制類除草劑的抗性[9-10]。目前在油菜基因組中已經發現的ALS基因有3個,即BnALS1、BnALS2、BnALS3[11]。有關油菜ALS基因的研究多集中在其抗除草劑的機制上,而關于ALS基因啟動子的研究及應用則鮮有報道。
本研究旨在克隆油菜ALS基因的啟動子區域序列,并通過轉基因瞬時表達驗證其啟動子活性。
1 材料與方法
1.1 材料
1.1.1 生物試劑 引物由南京擎科生物科技有限公司合成;限制性內切酶系NEB公司產品(NEB Ipswich,UK);高保真DNA聚合酶5×TransStart FastPfu PCR 試劑、凝膠回收試劑盒EasyPure?Quick Gel Extraction Kit、T/A克隆載體pEASY-T1Kit均為北京全式金生物技術有限公司產品;MS培養基系青島海博生物有限公司產品。
1.1.2 試驗材料 油菜材料為江蘇省農業科學院經濟作物研究所選育的抗除草劑品系M342;轉基因受體材料為本氏煙(Nicotianabenthamiana),由江蘇省農業科學院種質資源與生物技術研究所張保龍研究員提供。
1.2 試驗方法
1.2.1 啟動子序列克隆與啟動子分析 根據油菜的BnALS基因序列,通過美國國立生物技術信息中心(NCBI)網站(https://www.ncbi.nlm.nih.gov/)檢索其相應的油菜基因組序列,提取BnALS基因編碼區5′末端上游大約1 000個堿基的序列,通過專業網站(http://molbiol-tools.ca/Promoters.htm)進行啟動子預測分析。然后選取上游富含啟動子元件的1 000 bp左右的序列,設計特異性擴增引物,并添加限制性內切酶位點,在上游引物的5′末端添加限制性內切酶HindⅢ的酶切位點,在下游引物的5′末端添加NcoⅠ的酶切位點,引物為PF(5′-AAGCTT GTGGAGCTGATCTTACCGACCGAAC-3′)/PR(5′-CCATGGGGTTAGAGGAGAGAGAGATGATGAA-3′)。
1.2.2 啟動子序列擴增、克隆、測序及分析 采用十六烷基三甲基溴化銨(CTAB)法提取油菜M342葉片的基因組DNA。以此為模板,用高保真DNA聚合酶擴增油菜BnALS基因啟動子區域的堿基序列。擴增體系:10 μL 5×Trans Start FastPfu Buffer,1 μL基因組DNA,1 μL 10 μmol/L PF,1 μL 10 μmol/L PR,5 μL 2.5 mmol/L dNTPs,1 μL Trans Start FastPfu DNA聚合酶,用ddH2O補足總體積到50μL。PCR程序:95 ℃ 3 min,95 ℃ 30 s,55 ℃ 30 s,72 ℃ 1 min,35個循環;72 ℃,5 min。用瓊脂糖凝膠電泳檢查擴增產物,膠回收純化1 kb左右的PCR產物并克隆到T/A載體pEASY-T1上,轉化大腸桿菌DH5α,獲得重組質粒pEASY-P,挑取陽性克隆送南京擎科生物科技有限公司測序。
通過Plant CARE網站(http://bioinformatics.psb.ugent.be/webtools/plantcare /html/)對克隆到的油菜BnALS基因上游序列進行生物信息學分析。
1.2.3 啟動子功能驗證表達載體構建 將第1.2節克隆到的啟動子序列重組到pCAMBIA1304載體上GUS基因的5′端,這樣就構成了由BnALS基因啟動子驅動的1個完整的GUS基因表達框。分別提取克隆載體pEASY-P質粒和由筆者所在實驗室保存的植物表達載體pCAMBIA1304質粒,用限制性內切酶HindⅢ、NcoⅠ雙酶切質粒DNA,回收從pEASY-P質粒切下的大小約為1 000 bp的片段,通過連接酶的作用,將啟動子序列重組到目標載體pCAMBIA1304的相應酶切位點上。雙酶切反應體系:總體積為50 μL,其中含有5 μL緩沖液、30 μL質粒DNA溶液、2 μLHindⅢ、2 μLNcoⅠ、16 μL H2O,于37 ℃放置6 h或過夜。酶切后,通過瓊脂糖凝膠電泳,回收相應的片段,進行連接反應,連接酶反應體系:總體積為20 μL,含有5 μL線性化載體DNA、5 μL啟動子DNA溶液、2 μL T4連接酶緩沖液、2 μL T4連接酶、6 μL H2O,于 16 ℃ 過夜。將連接反應產物用熱激法轉化大腸桿菌感受態細胞DH5α,在含有卡那霉素(Kan)的LB培養基上于37 ℃培養過夜。
1.2.4 煙草轉基因試驗 為了進一步驗證所克隆啟動子的生物學功能,將構建的表達載體pCAMBIA1304-P轉入農桿菌EHA105,利用葉盤轉化法轉染煙草。具體試驗過程如下:
1.2.5 農桿菌的準備 挑取攜帶載體pCAMBIA1304-P的農桿菌EHA105的單菌落,接種于5 mL含有20 mg/L利福平(Rif)和50 mg/L Kan的LB液體培養基中,28 ℃振蕩培養過夜。取活化過夜的農桿菌,按1 ∶50的比例稀釋到含有 20 mg/L Rif和50 mg/L Kan的YEB液體培養基中,繼續培養至D600 nm約為0.6~0.8。于6 000 r/min離心5 min,收集菌體,用1/2 MS液體培養基洗滌1次菌體,并將其稀釋至D600 nm為0.30~0.35。
1.2.6 煙草葉盤的遺傳轉化 選取苗齡約為30 d的煙草無菌苗,在無菌條件下用手術刀切取大小約為0.8 cm2的煙草葉片為外植體,投入已經準備好的農桿菌菌液中,振蕩侵染 5 min 后,取出并用濾紙吸干附著于葉片表面的殘液,然后放在共培養基(1×MS,3%蔗糖,1%瓊脂粉,2.0 mg/L BA,0.5 mg/L IAA,pH值為5.8)上的暗處,溫度設為25 ℃,共培養48 h。
1.2.7 轉基因煙草苗的再生 將共培養后的煙草外植體轉到芽誘導培養基(配方為1×MS+3%蔗糖+2.0 mg/L BA+0.5 mg/L IAA+500 mg/L羧芐青霉素+20 mg/L潮霉素+1%瓊脂粉,pH值為5.8)上進行芽誘導培養,每隔2~3周繼代1次。繼代用的培養基同芽誘導培養基。待不定芽長出后,且芽長為1.0~1.5 cm時,將芽切下換到生根培養基(配方為1×MS+3%蔗糖+0.5 mg/L IAA+500 mg/L羧芐青霉素+20 mg/L潮霉素+1%瓊脂粉,pH值為5.8)上誘導生根。
1.2.8 轉基因煙草植株的PCR鑒定 取轉基因處理的再生煙草苗葉片,用CTAB法提取基因組DNA,另取非轉基因煙草苗葉片DNA作為陰性對照。用特異性引物進行PCR鑒定。引物序列:PGF,5′-CGTTCACAAACTCATTCATCATCTC-3′;PGR,5′-TTTGATGCCGTTCTTTTGCTTGTCG-3′。20 μL 擴增反應體系:10 μL 2×Tag master mix,1 μL DNA模板,1 μL 引物PGF,1 μL引物PGR,7 μL H2O。反應程序:94 ℃ 4 min,94 ℃ 40 s,55 ℃ 40 s,72 ℃ 40 min,35個循環;72 ℃ 5 min,用瓊脂糖凝膠電泳檢測擴增產物。
1.2.9 GUS活性檢測 用北京Solarbio生物科技有限公司生產的GUS染色液試劑盒定性檢測GUS基因的表達活性。將轉基因煙草PCR陽性單株幼苗完整取出,去除根部的瓊脂,用無菌水清洗后,將植株浸泡在GUS顯色液中,于室溫下放置3~6 h后,用無水乙醇進行脫色處理,鏡檢拍照。以非轉基因幼苗作為對照。
2 結果與分析
2.1 油菜ALS基因啟動子序列的克隆與分析
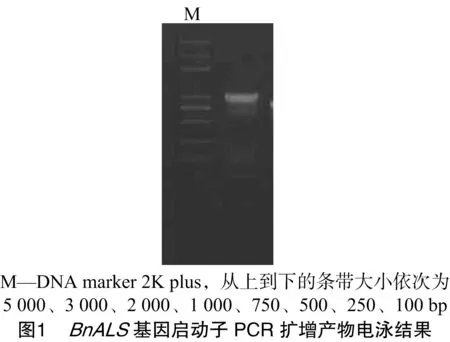
本研究根據BnALS基因上游序列設計特異性引物,通過PCR擴增油菜基因組DNA的方法獲得大小約為1 000 bp的片段,詳見圖1。對該片段進一步克隆、測序,實際獲得長度為1 048 bp的BnALS基因上游序列(序列1),詳見圖2。
通過Plant CARE網站(http://bioinformatics.psb. ugent.be/webtools/plantcare/html/)對克隆到的BnALS基因上游序列進行生物信息學分析,結果顯示,該序列含有與啟動子相關的多種順式作用元件,如CAAT-box、TATA-box等,表明該序列具有真核細胞啟動子的結構特征。部分順式作用元件的情況見表1。
2.2 啟動子功能驗證表達載體的構建
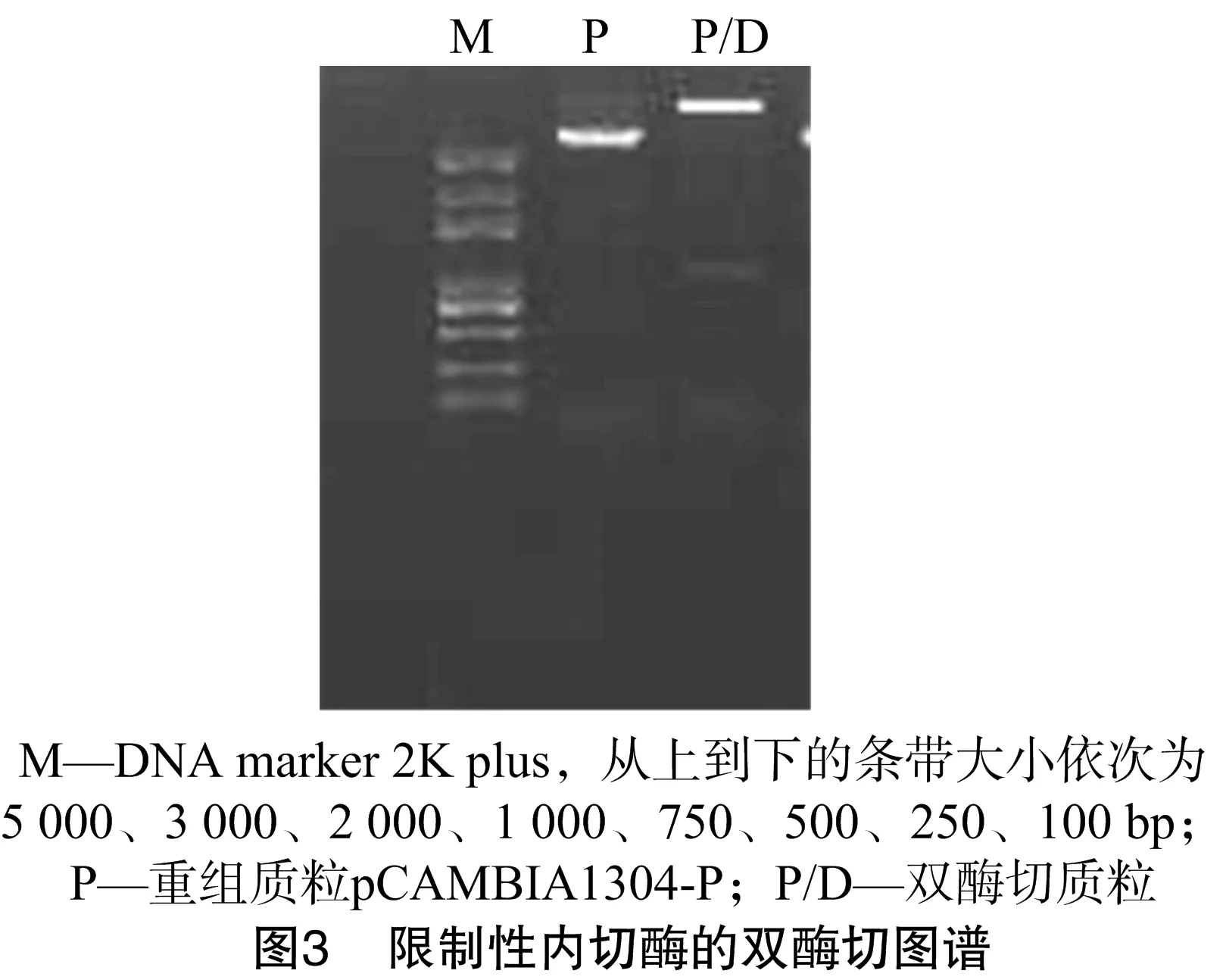
為了進一步驗證所克隆的BnASL基因上游序列是否具有啟動子功能,將該序列重組到pCAMBIA1304載體上GUS基因的5′-端,這樣就構成了由BnALS基因的上游序列驅動的1個完整的GUS基因表達框。分別提取獲得的克隆載體pEASY-P質粒和筆者所在實驗室保存的植物表達載體pCAMBIA1304質粒,并用限制性內切酶HindⅢ、NcoⅠ雙酶切質粒DNA,回收從pEASY-P質粒上切下的大小約為 1 000 bp 的片段,重組到雙酶切線性化的pCAMBIA1304載體上,獲得重組載體pCAMBIA1304-P。重組載體經限制性內切酶酶切驗證,可以得到1個大小約為1kb的片段,證明啟動子序列已經重組到目標載體上(圖3)。經測序分析可知,所克隆的啟動子序列準確插入相應的酶切位點,無缺失、錯配現象,表明該重組載體可以用于轉基因功能的驗證工作。


表1 ALS啟動子序列含有的主要相關順式作用元件
2.3 煙草轉基因試驗
為了進一步驗證所克隆的油菜BnALS基因上游序列的啟動子功能,將構建的表達載體pCAMBIA1304-P轉入農桿菌EHA105,利用葉盤轉化法轉染煙草。煙草葉片經過農桿菌侵染、抗性芽誘導、抗性芽生根等階段的再生培養,最終獲得轉基因煙草幼苗,其過程詳見圖4。
2.4 轉基因煙草植株的PCR鑒定
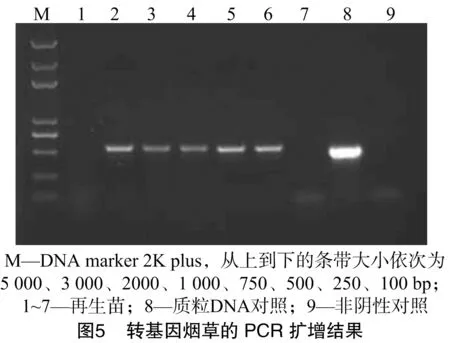
利用設計的特異性PCR引物擴增再生幼苗的基因組DNA,鑒定轉基因陽性單株。由圖5可以看出,經過PCR檢測,有5株幼苗擴增到與質粒對照相同的陽性條帶,2株幼苗未擴增到相應的條帶。

2.5 GUS基因表達活性檢測
以PCR結果為陽性的轉基因再生煙草幼苗及非轉基因處理的煙草幼苗對照,用GUS顯色試劑盒進行GUS基因表達活性的定性檢測。將煙草幼苗完整取出,浸泡在GUS基因顯色液中,于室溫下放置3~6 h后,用無水乙醇脫除背景色,如果有GUS基因表達活性,組織就會出現藍色反應。結果顯示,轉基因煙草全株均出現明顯的藍色反應,而非轉基因幼苗則呈淡淡的黃色,轉基因煙草GUS活性定性檢測結果見圖6。以上結果表明,本研究克隆的BnALS基因上游序列具有較強的組成型表達活性, 能驅動報告基因在煙草幼苗各個器官中的表達。

3 討論
與其他真核生物一樣,植物啟動子區域最具特征的就是TATA box序列,這是RNA聚合酶Ⅱ識別的位點,也是一些反式作用因子與DNA相互作用的位點之一。TATA box與轉錄起始點之間的堿基長度是轉錄精確起始的必需因素,植物啟動子的TATA box多在轉錄起始點上游(32±7) bp處[12]。TATA box通過與轉錄因子TFⅡD的識別結合而發揮功能,決定RNA聚合酶起始轉錄的位點,以及介導前轉錄復合物的形成并起始轉錄;起始因子(initiator,簡稱Inr)是基因啟動子核心結構的第2種類型,與轉錄起始位點重疊[13]。Inr元件并無十分嚴格的同源順序,其中最關鍵的核苷酸是處于+1位置的A和+3位置的T。Inr在功能上與TATA框類似,常通過與TFIID的結合決定轉錄起始點的位置起始轉錄,并能介導上游至少一部分激活因子的調控作用;CAAT box是Shirsat等于1989年在豌豆legA基因啟動子中發現的1個增強轉錄的元件,一般位于-75 bp附近,其保守序列為GGC/TCAATCT[14]。CAAT box控制著轉錄起始的頻率,其對基因轉錄的激活作用存在雙向性,且作用距離不固定;GC box通常位于-90 bp附近,保守序列為GGGCGG,可有多個拷貝,并能以任何方向存在而不影響其功能。Kuwahara等研究發現,GC box需要與另一特異的轉錄因子(SP1)結合才能促進基因轉錄[15]。植物啟動子的順式元件除了CAAT box和GC box以外,不同來源的啟動子通常還具有種屬特異的或者僅局限于某種基因家族特有的調節序列。
本試驗根據油菜BnALS基因上游序列設計特異性引物,通過PCR擴增獲得的ALS基因上游的1 048個堿基序列中包含多個TATA box和CAAT-box啟動子核心序列,僅在正義鏈上就檢測到11處TATA box序列和3個CAAT-box,這些啟動子的核心序列可能決定了轉錄的起始、方向和轉錄效率。根據植物基因轉錄起始位點通常與上游TATA-box 的距離為(32±7) bp,以及轉錄起始堿基多為A堿基的規律,推測本試驗克隆的啟動子起始位點可能是位于1 026 bp或 1 028 bp 處的A堿基。此外,BnALS基因上游區域還含有其他多種轉錄調控元件,主要有激素響應元件、脫落酸響應元件ABRE-motif、甲基茉莉酸響應元件CGTCA-motif和生長素等激素響應元件TGA-element,說明ALS基因在激素響應過程中可能起作用。此外,還有非生物脅迫響應元件HSE和MBS,因此可見,ALS基因也可能參與干旱或熱激反應的應答。另外,還發現有多個光響應元件,暗示ALS基因的表達可能還受到光的調控。
人們研究啟動子的目的主要是為了弄清一些結構基因的表達與調控模式,除此以外,啟動子在植物基因工程研究領域也有重要的應用價值。啟動子對外源基因的表達水平影響很大,是基因工程表達載體的重要元件。目前在植物基因工程中應用最多的啟動子是來自花椰菜花葉病毒(CaMV)的35S啟動子和農桿菌胭脂堿合酶基因的nos啟動子[16]。這2個組成型表達的啟動子常用于雙子葉植物基因工程,而在單子葉植物中應用較多的是來自玉米的Ubi1啟動子[17]和水稻的Act1啟動子[18]。人們不斷開發研究新啟動子應用于植物的基因工程,目的是為了在植物中更高效地表達目的基因。重復使用同一種組成型啟動子驅動2個或2個以上的外源基因表達可能會引起基因沉默或者發生共抑制[19]。另外,將從病毒基因組中克隆的啟動子序列應用到植物基因工程中,可能會引起人們對轉基因植物生物安全性的擔憂,因此,從植物本身克隆活性強的組成型啟動子也就成了植物基因工程研究的一個重要方面。Shirasawa-Seo等從擬南芥中克隆了色氨酸合成酶亞基(PTSB1)和植物光敏色素B基因的啟動子(PPHYB),替代CaMV35S的組成型啟動子,二者分別與GUS基因融合表達,活性甚至高于35S啟動子,驅動NPTⅡ基因表達,具有相同的效果,可以認為擬南芥的PTSB1和PPHYB啟動子是植物來源的可替代35S啟動子的強組成型啟動子[20]。本試驗克隆到了BnALS基因啟動子序列,通過生物信息學軟件預測了啟動子元件組成,并構建了與GUS基因融合的植物表達載體,通過轉基因煙草的瞬間表達分析,初步結果顯示,所克隆的啟動子序列能驅動GUS基因在煙草幼苗多個器官中表達,表明該序列具有較強的組成型啟動子的表達活性。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
少兒科學周刊·兒童版(2017年5期)2017-06-29 22:24:28
少兒科學周刊·兒童版(2017年5期)2017-06-29 16:46:33
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
當代化工研究(2016年9期)2016-03-20 16:22:08
少兒科學周刊·兒童版(2015年10期)2015-11-07 03:42:03
少兒科學周刊·兒童版(2015年1期)2015-07-07 04:12:52
聲屏世界(2014年6期)2014-02-28 15:18:09
