復(fù)方楓蓼顆粒制備工藝研究
2019-02-26 00:56:08高彪蔣琪向蓉陳文露徐志宏彭新宇
中國獸藥雜志 2019年1期
高彪,蔣琪,向蓉,陳文露,徐志宏,彭新宇
(廣東省農(nóng)業(yè)科學(xué)院動物衛(wèi)生研究所 農(nóng)業(yè)部獸用藥物與診斷技術(shù)廣東科學(xué)觀測實驗站 廣東省畜禽疫病防治研究重點實驗室 廣東省中獸藥工程技術(shù)研究中心, 廣州 510640)
復(fù)方楓蓼是民間防治胃腸道疾病的經(jīng)典中藥復(fù)方。由牛耳楓和辣蓼兩味中藥按2∶1的比例組成[1]。牛耳楓(DaphniphyllumcalycinumBenth.)又稱牛耳鈴,是屬虎皮楠科(Daphniphyllaceae)虎皮楠屬植物,具有極高的藥用價值,果實及其枝葉均可入藥[2]。該藥具有活血散瘀、祛風(fēng)止痛、止痢清熱等功效,主治痢疾、風(fēng)濕骨痛、感冒發(fā)熱等疾病[3]。辣蓼(polygonumhydropiperL)又稱柳蓼,為蓼科(Polygonaceae)蓼屬(polygonum)植物[4]。全草可入藥,具有止痢、活血化瘀、止癢殺蟲等功效。主治腹瀉、腸胃炎、風(fēng)濕性關(guān)節(jié)痛、跌打損傷等[5]。現(xiàn)代藥理研究表明,該復(fù)方中含有黃酮類、倍半萜類、生物堿類、有機(jī)酸類等化學(xué)成分[6-7],具有抗炎、鎮(zhèn)痛、抑菌、抗?jié)冃越Y(jié)腸炎、保護(hù)胃黏膜等多方面的藥理作用,臨床用其治療急性胃腸炎、食滯胃痛等癥,具有良好效果[8-9]。
牛耳楓、辣蓼臨床用于預(yù)防及治療胃腸道時,多為湯劑。但是湯劑煎煮程序麻煩、口服量大、口味不佳,且攜帶、貯存不變,與現(xiàn)代生活不相符合。而顆粒劑作為一種固體制劑,攜帶、貯藏及服用均方便,且藥物穩(wěn)定性及生物利用度高。研究通過單因素和正交實驗確定了最佳提取工藝。在此基礎(chǔ)上采用濕法制粒,考察顆粒劑的制備方法,以吸濕率、粒度合格率及溶化性、外觀為考察指標(biāo)篩選出楓蓼顆粒劑的制備工藝,并進(jìn)行半成品的質(zhì)量控制。旨在研究出質(zhì)量穩(wěn)定,服用方便的復(fù)方楓蓼顆粒。
1 材料與方法
1.1 材料 高效液相色譜儀,美國Waters公司;RE-5399型旋轉(zhuǎn)蒸發(fā)儀,上海亞榮生化儀器廠;UV-3100PC型紫外分光光度計,上海美譜達(dá)儀器有限公司;SB25-12DT型超聲波清洗機(jī),寧波新芝生物科技股份有限公司;Metter-Toledo-xs型十萬分之一天平,瑞士梅特勒公司;SHZD-D(Ⅲ)型循環(huán)式真空泵,鞏義市予華儀器有限責(zé)任公司;804R型低溫離心機(jī),上海艾本德生物技術(shù)有限公司;DZF-6050型真空干燥箱,上海申賢恒溫設(shè)備廠;HWS-158型恒溫恒濕箱,寧波江南儀器廠;101-2型電熱鼓風(fēng)干燥器,上海浦東躍欣科學(xué)儀器廠。
牛耳楓藥材購于廣東茂名(批次201507);辣蓼藥材購于廣東茂名(批次201508);經(jīng)廣東藥學(xué)院李書淵教授鑒定為蓼科植物水辣蓼PolygonumhydropiperLinn.和虎皮楠科植物牛耳楓DaphniphyllumcalycinumBench.。藥材經(jīng)低溫干燥后,粉碎,過20目篩,按牛耳楓辣蓼2∶1的比例混合均勻,備用。
蘆丁,批號100080-201409,中國食品藥品檢定研究院,純度≥98%;槲皮素,批號10081-201509,中國食品藥品檢定研究院,純度≥98%;乙腈,色譜純;超純水,自制;糊精;蔗糖;可溶性淀粉及實驗中所用的其他試劑均為分析純。
1.2 方法
1.2.1 提取液的配制 取楓蓼藥粉(牛耳楓∶辣蓼=2∶1)20g,精密稱定,置于500 mL圓底燒瓶中,按不同的提取工藝對藥物進(jìn)行回流提取,過濾,減壓濃縮,溶劑定容至100 mL。
1.2.2 總黃酮含量的測定[16]精密吸取提取液1 mL,加30%乙醇至6 mL,加5%NaNO2溶液1 mL,搖勻,放置6 min;加10%Al(NO3)3溶液1 mL,搖勻,放置6 min;加4.3%NaOH溶液10 mL,再加30%乙醇溶液定容至刻度,搖勻,放置15 min后,以相應(yīng)試劑為空白對照,于510 nm處依照UV法測定吸光度,通過總黃酮標(biāo)準(zhǔn)曲線方程計算總黃酮含量。
1.2.3 槲皮素含量的測定[17]色譜條件:Waters Symmetry shield RP18 色譜柱(4.6 mm×250 mm,5.0 μm);流動相:A為乙腈,B為0.4%磷酸水溶液;梯度洗脫:0~13 min,A為20%~45%;13~15 min,A為45%~60%;15~20 min,A為70%~20%;流速:1 mL/min;檢測波長:370 nm;柱溫:30 ℃;進(jìn)樣量10 μL,理論塔板數(shù)按槲皮素峰計算應(yīng)不低于3000。測定槲皮素峰面積,通過標(biāo)準(zhǔn)曲線方程計算槲皮素含量。
1.2.4 出膏率的測定 精密量取定容后的楓蓼藥液50 mL,置于已經(jīng)干燥至恒定重量的蒸發(fā)皿中,將其水浴蒸干后,105 ℃恒溫干燥3 h后,置于干燥器中放冷0.5 h后,迅速稱重,計算出膏率。
1.2.5 提取工藝優(yōu)化
1.2.5.1 提取溶劑的考察 水提取法:取楓蓼藥粉20 g,精密稱定,置于圓底燒瓶中,加水200 mL,浸泡1 h,加熱回流1 h,減壓濃縮至100 mL,過濾,即得,測定槲皮素、總黃酮含量及出膏率。
乙醇提取法:取楓蓼藥粉20 g,精密稱定,置于圓底燒瓶中,分別加45%、65%、85%的乙醇200 mL、浸泡1 h,減壓濃縮至100 mL,過濾,即得,測定槲皮素、總黃酮含量及出膏率。
1.2.5.2 正交試驗 在提取溶劑考察的基礎(chǔ)上,選擇料液比、乙醇濃度、提取時間及提取次數(shù)為影響提取效率的主要因素,設(shè)計三個水平,采用正交試驗,見表1,以總黃酮、槲皮素的含量及出膏率為考察指標(biāo),進(jìn)行綜合評分,優(yōu)選楓蓼藥材最佳提取工藝。
1.2.6 濃縮干燥工藝的研究
1.2.6.1 濃縮工藝 減壓濃縮具有濃縮效率高,溫度低、有效成分損失少等特點。故本節(jié)對濃縮溫度進(jìn)行考察。按最佳提取工藝平行提取9次,制備9份提取液,6000 r/min離心10 min后,分別在0.08 mpa下以50 ℃、60 ℃、70 ℃濃縮至相對密度為1.15~1.25的浸膏,測定總黃酮平均含量。考察最佳濃縮工藝。
1.2.6.2 減壓干燥工藝 溫度是影響干燥效率的主要因素。故本節(jié)采用單因素變量法重點討論溫度的影響。將相對密度為1.15~1.25的浸膏分成9份,每份100 g,于60 ℃、70 ℃、80 ℃下真空干燥,以總黃酮含量為指標(biāo),考察最佳減壓工藝。
1.2.7 成型工藝
1.2.7.1 輔料的選擇 以制粒難易程度,外觀、硬度等作為指標(biāo),篩選糊精、蔗糖、可溶性淀粉等輔料,找出最佳比例,確定濕法制粒最佳工藝條件。
1.2.7.2 濕法制粒 預(yù)實驗表明影響制粒的因素主要包括:干燥溫度、乙醇濃度、輔料用量及輔料與藥粉的比例。故采用四因素三水平L9 (34)正交表(表2)安排正交試驗,以吸濕率、粒度合格率、外觀溶化性為考察指標(biāo)進(jìn)行處方篩選。
取已制備好的干膏粉30 g,以14目篩制顆粒,干燥后取出放冷至室溫,14目篩整粒,即得。
吸濕率 精密稱定楓蓼顆粒2 g,置于干燥至恒定重量的稱量瓶底部,將稱量瓶放置在相對濕度為75%干燥器內(nèi)(瓶蓋已打開)25℃放置48 h后稱定顆粒重量。按公式(2)計算吸濕率。
吸濕率=(吸濕后顆粒重量—吸濕前顆粒重量)/吸濕前顆粒劑重量 ×100%(2)
粒度合格率 按照2010年版《中國藥典》一部附錄XIB法,取顆粒50 g于一號篩中,下面放置五號篩。水平往返輕叩3 min,取合格顆粒稱重并按公式(3)計算粒度合格率。
粒度合格率(%)=合格的顆粒重量/過篩前重量×100%(3)
外觀溶化性 外觀根據(jù)顆粒硬度、均勻度及色澤進(jìn)行判斷;溶化性按照2010年版《中國藥典》一部附錄ⅠC項下溶化性檢查法進(jìn)行考察。二者共同采用十分制進(jìn)行打分。
1.2.5.3 提取工藝驗證試驗 取楓蓼藥粉3份,每份10 g,按照最佳提取工藝,以10倍量65%的乙醇回流提取2次,每次60 min,收集合并提取液,過濾,測定總黃酮含量、槲皮素含量及出膏率,計算RSD,對最佳提取工藝進(jìn)行驗證。
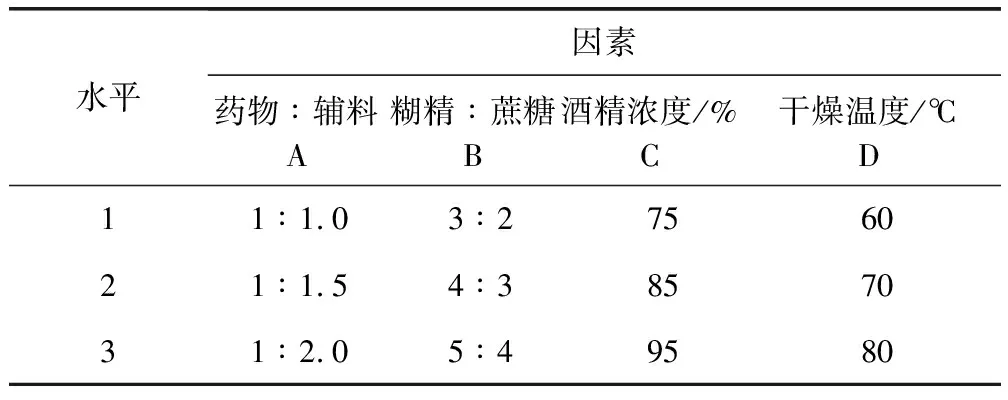
表2 制劑成型工藝因素水平表Tab 2 Factors of level of orthogonal test
1.2.7.3 制備工藝驗證試驗 按優(yōu)選出的最佳工藝進(jìn)行重復(fù)性試驗驗證。
1.2.8 半成品質(zhì)量控制 為了保證成品顆粒制劑的穩(wěn)定性及一致性,在制劑的制備過程中需要對顆粒劑半成品進(jìn)行質(zhì)量控制,故本研究考察了溶化性、休止角及臨界相對濕度。
1.2.8.1 溶化性 照2010年版《中國藥典》一部附錄ⅠC項下溶化性檢查法進(jìn)行考察。5 min內(nèi),三批樣品全部溶解,達(dá)到藥典標(biāo)準(zhǔn)。
1.2.8.2 休止角 休止角是考察粉體或顆粒流動性的指標(biāo),通常采用漏斗法測定其角度。將漏斗置繪圖紙上方一定的距離H,把制備好的顆粒從漏斗中倒入,直至顆粒形成的圓錐頂端與漏斗的出口處接觸,圓錐體的直徑為2R,錐體高度H,根據(jù)公式tanα=H/R即可求出休止角α,α越小流動性越好。
1.2.8.3 臨界相對濕度 為了確定環(huán)境濕度對顆粒制劑生產(chǎn)及貯存的影響,故本節(jié)需對顆粒的臨界相對濕度作出考察。
取干燥至恒重的成品顆粒2 g,平行取三份,放入相對濕度為20%、30%、40%、50%、60%、70%、80%、90%的環(huán)境中,25℃下放置96 h后取出,測定吸濕前后顆粒重量變化。
吸濕率=(吸濕后顆粒重量—吸濕前顆粒重量)/吸濕前顆粒重量×100% (4)
1.2.9 數(shù)據(jù)處理 采用SPSS20. 0 統(tǒng)計軟件對所得所有數(shù)據(jù)進(jìn)行處理,正交試驗結(jié)果進(jìn)行單因素方差分析。
2 結(jié)果與分析
2.1 提取溶劑的考察結(jié)果 分別考察了水回流提取以及45%、65%、85%乙醇提取的提取效率,結(jié)果見表3。以乙醇為提取溶劑時的提取效率優(yōu)于以水為提取溶劑時的提取效率。故選擇乙醇作為提取溶劑。
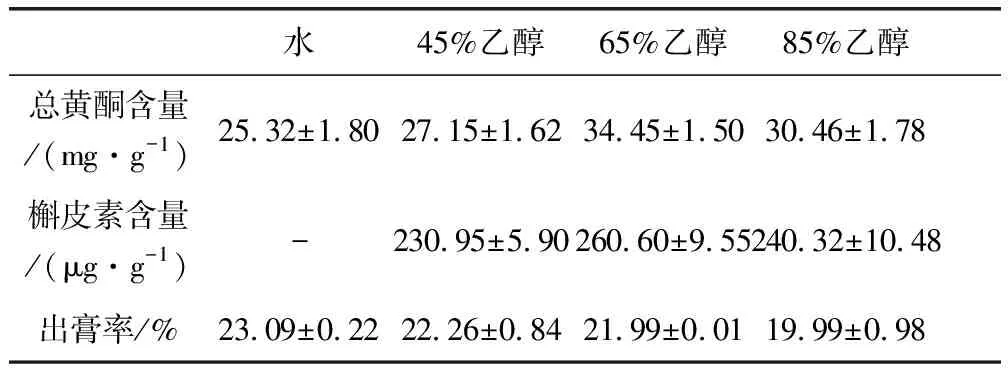
表3 不同提取溶劑試驗結(jié)果Tab 3 Different extraction solvent tests
2.2 方差分析結(jié)果 由正交試驗和方差分析結(jié)果(表4和表5)可知,A、B因素對提取效果具有顯著性意義(P<0.05),C因素差異不顯著,極差A(yù)>B>C>D,故對實驗結(jié)果影響主次順序為A(乙醇濃度)>B(提取次數(shù))>C(提取時間)>D(加醇量),A因素中A2>A3>A1,B因素中B2>B3>B1,C、D為次要因素,故從節(jié)約時間原則上確定最優(yōu)綜合評分方案為A2B2C1D2。綜合以上分析,確定本次試驗最佳工藝為:10倍量65%的乙醇回流提取2次,每次60 min。
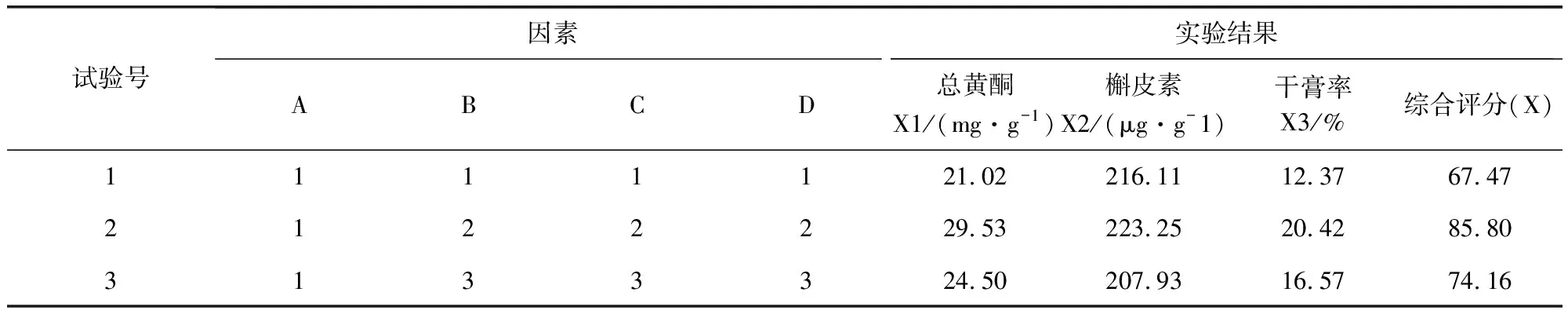
表4 正交試驗結(jié)果表Tab 4 The results of the orthogonal
續(xù)表
X=(X1/X1mas)×40+(X2/X2mas)×40+(X3/X3mas)×20
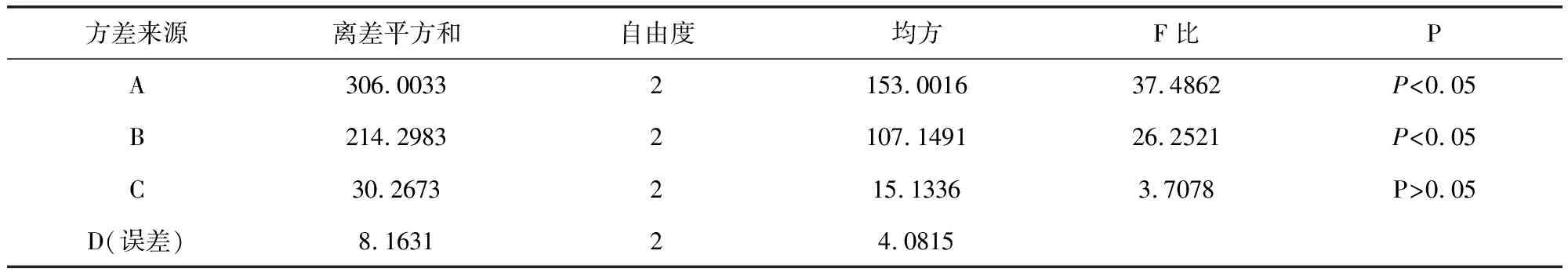
表5 醇提取結(jié)果方差分析表Tab 5 Ethanol extraction of variance results
F0.05(2,2)=19
2.3 提取工藝驗證結(jié)果 由表6可知,總黃酮RSD為0.88%、槲皮素RSD為0.14%,出膏率RSD為1.85%,說明該法重復(fù)性好。
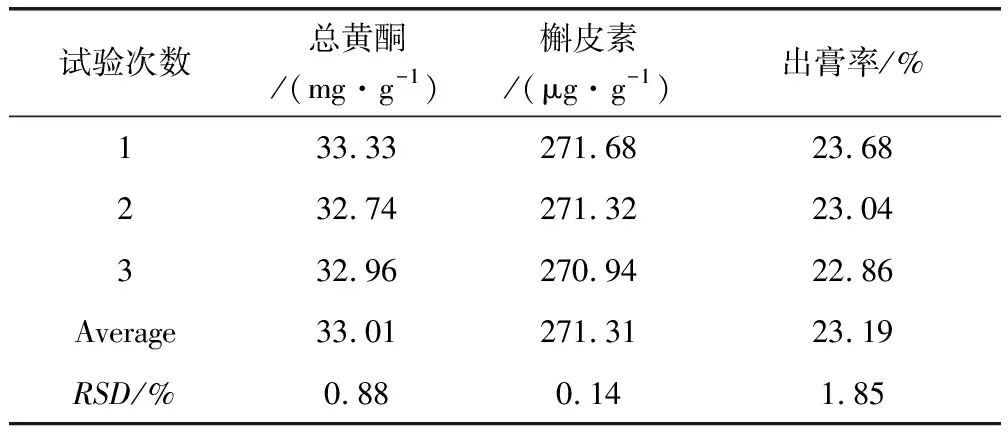
表6 提取工藝驗證結(jié)果表Tab 6 Confirming test of the technology
2.4 濃縮干燥工藝結(jié)果
2.4.1 濃縮工藝結(jié)果 將相對密度為1.15~1.25的浸膏分成9份,每份100 g,于60 ℃、70 ℃、80 ℃、90 ℃下真空干燥,計算干燥后總黃酮含量,由表7可知50 ℃時總黃酮含量最高,70 ℃時總黃酮含量最低,50 ℃和60 ℃含量差別不大,而70 ℃和60 ℃含量差別相對較大,因溫度越高,濃縮時間越短,故選擇濃縮溫度為60 ℃減壓濃縮。
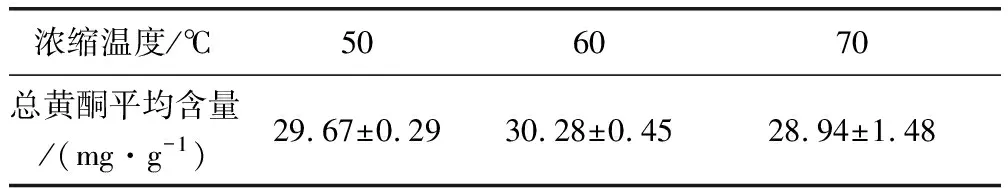
表7 濃縮試驗結(jié)果表Tab 7 The results of concentrate test
2.4.2 減壓干燥工藝結(jié)果 由表8可知70 ℃總黃酮含量最高,80 ℃時次之,90 ℃最低,可能原因是60 ℃干燥時間過久,90 ℃溫度過高導(dǎo)致成分損失。80 ℃相比之下干燥時間斷,成分損失相對較少,故選擇干燥溫度為80 ℃。
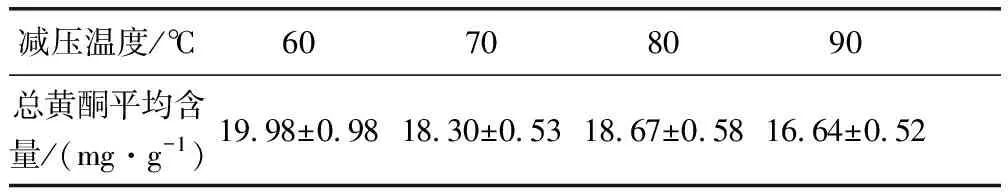
表8 減壓干燥試驗結(jié)果表Tab 8 The results of decompression drying
2.5 成型工藝
2.5.1 輔料的選擇結(jié)果 對于顆粒劑來說,目前常用的賦形劑包括乳糖、可溶性淀粉、蔗糖、糊精等。常用的潤濕劑包括水和不同濃度的乙醇。預(yù)實驗中考察了糊精、可溶性淀粉、蔗糖的吸水性。由圖1知,糊精與蔗糖吸濕性較低。糊精具有價格低廉,且易溶于水,成型性好等特點,蔗糖用于藥用輔料時具有矯味及粘合的作用。綜合制粒效果及成本方面的考慮,故決定采用糊精及蔗糖組成混合的輔料。
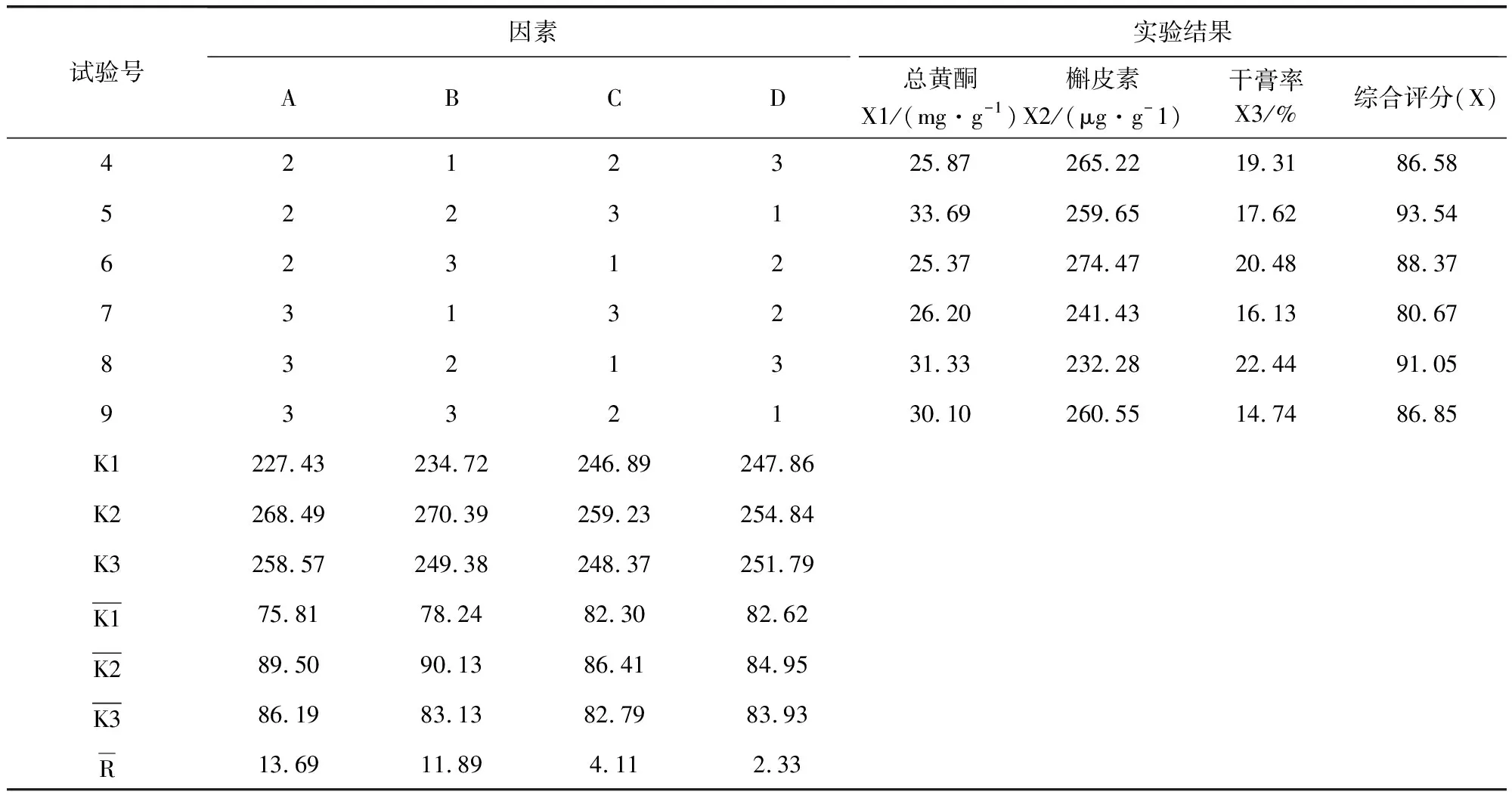
2.5.2 濕法制粒 以干燥溫度,乙醇濃度,輔料用量及輔料與藥粉的比例為考察因素,選擇三個水平,以吸濕率、粒度合格率,外觀溶化性作為考察指標(biāo)進(jìn)行處方篩選,根據(jù)正交試驗結(jié)果表(表9)及方差分析表(表10-表12)可知,在吸濕率測定中,C因素對試驗結(jié)果具有顯著性影響,A、B因素影響不顯著,極差C>B>A>D,故影響吸濕率的主次順序是C(酒精濃度)>B(糊精:蔗糖)>A(藥物:輔料)>D(溫度),C因素中C2>C1>C3,最優(yōu)組合為處A3B1C2D1 。 在粒度合格率測定中,A因素對試驗結(jié)果具有顯著性影響,B、C因素影響不顯著,極差A(yù)>B>C>D,故影響吸濕率的主次順序是A(藥物:輔料)>B(糊精:蔗糖)>C(酒精濃度)>D(溫度),A因素中C2>C1>C3,最優(yōu)組合為處A2B2C2D1。 在溶化性、外觀測定中,A、B、C、D各因素差異均不顯著,所以根據(jù)極差A(yù)>C>B=D,最優(yōu)組合為A2B3C1D1。由表13綜合分析可知,優(yōu)選工藝為A2B1C2D1。即85%乙醇作潤濕劑,藥物與輔料比例為1∶1.5,糊精與蔗糖比例為3∶2,干燥溫度為60 ℃。
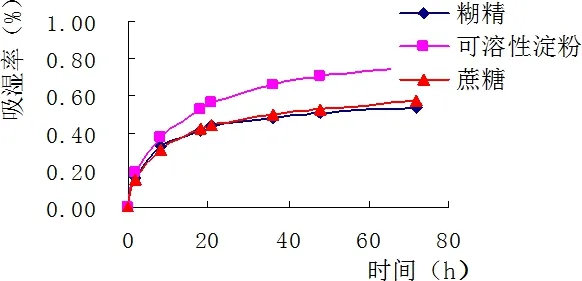
圖1 輔料吸濕性考察Fig 1 Hygroscopicity test
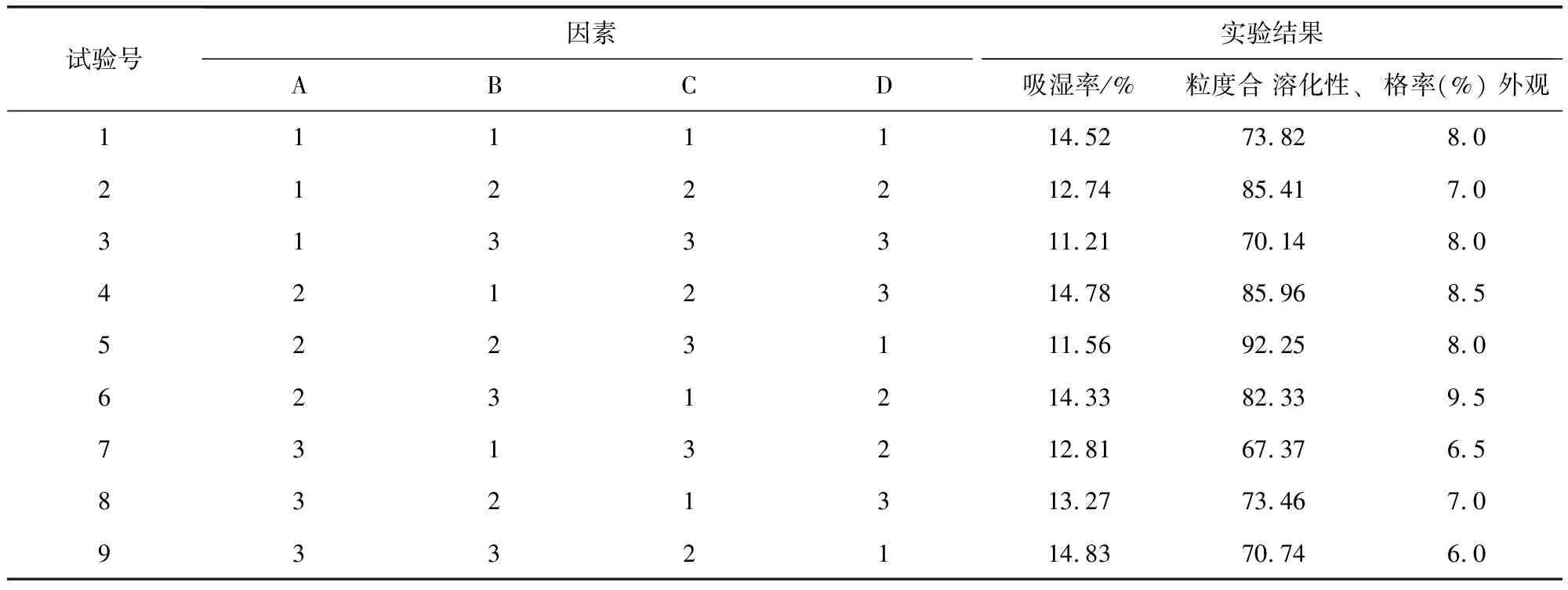
表9 制劑成型工藝正交試驗結(jié)果表Tab 9 Results of orthogonal test
續(xù)表
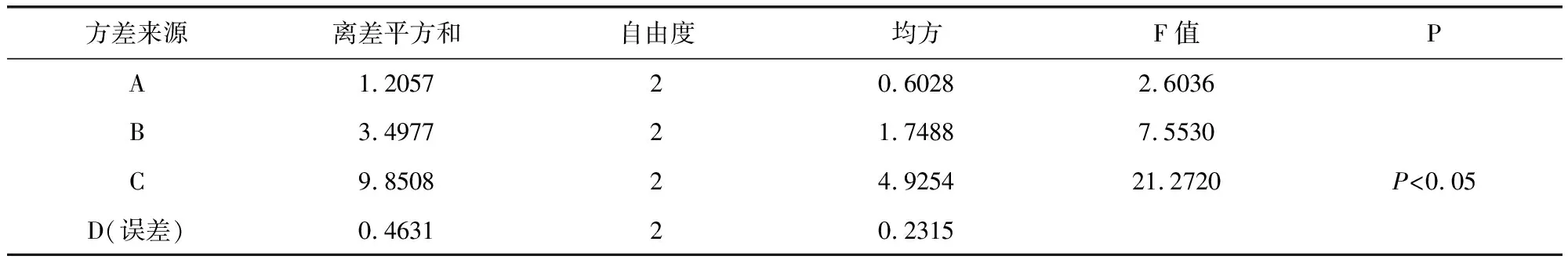
表10 吸濕率方差分析表Tab 10 Hygroscopicity of variance results
F0.05(2,2)=19
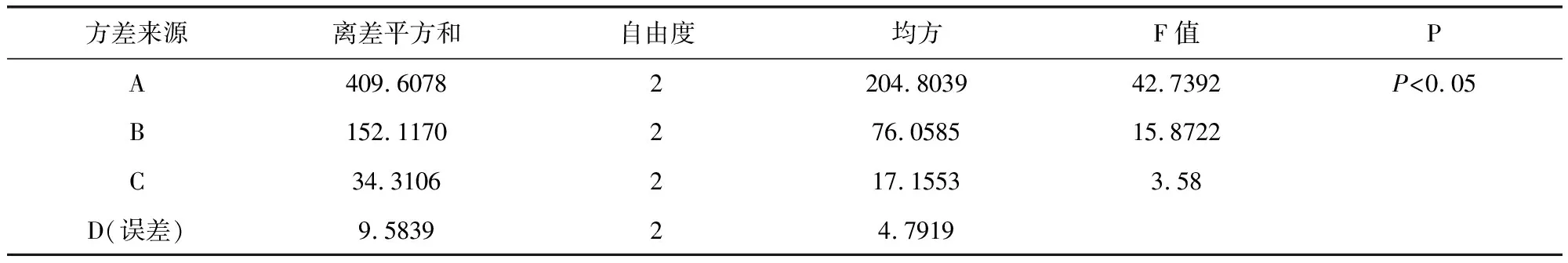
表11 粒度合格率方差分析表Tab 11 Granule pass rate of variance results
F0.05(2,2)=19
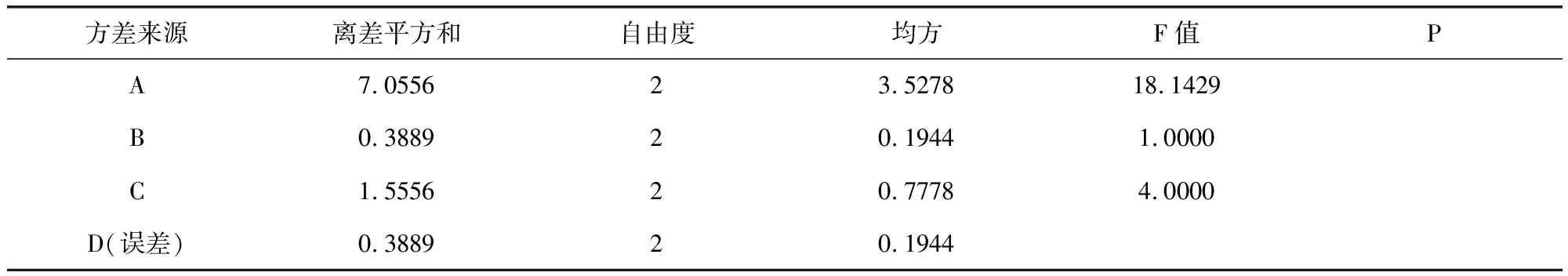
表12 溶化性、外觀方差分析表Tab 12 Solubility and exterior of variance results
F0.05(2,2)=19

表13 工藝優(yōu)化綜合分析表Tab 13 The results of process optimization
2.5.3 制備工藝驗證試驗 由表14可知,吸濕率RSD為1.72%,粒度合格率RSD為90.99%,溶化性、外觀RSD為0%,制備工藝重復(fù)性好。
2.6 半成品質(zhì)量控制結(jié)果
2.6.1 溶化性 照2010版本《中國藥典》一部附錄ⅠC項下溶化性檢查法進(jìn)行考察。5 min內(nèi),三批樣品全部溶解,達(dá)到藥典標(biāo)準(zhǔn)。
2.6.2 休止角 為錐體直徑為2R,錐體高度為H,tanα=H/R,計算α。由表13可知:平均休止角為34.2°<40°,表明顆粒流動性較好。

表14 驗證試驗結(jié)果表Tab 14 Verification of moulding technology

表15 止角試驗結(jié)果表Tab 15 Angle of repose granule
2.6.3 臨界相對濕度 不同濕度條件下顆粒平均吸濕率見表16。以環(huán)境相對濕度RH%為橫坐標(biāo),平均吸濕率(%)為縱坐標(biāo)作圖。 由圖2知,臨界相對濕度在67%以上時,吸濕率明顯加大。因此在制粒、包裝、貯藏時,外界濕度應(yīng)控制在67%以下。

表16 顆粒吸濕平衡試驗結(jié)果表Tab 16 Moisture absorption rate of granule
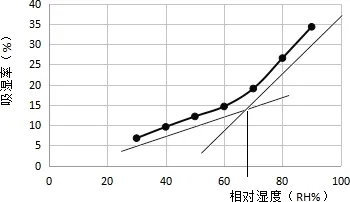
圖2 不同相對濕度下浸膏粉的吸收率Fig 2 Hygroscopic rates in different RH
3 討論與結(jié)論
中藥顆粒劑是指中藥材經(jīng)過不同溶劑的提取,再與適宜的輔料或不封藥材細(xì)粉以一定比例混勻,制成的感顆粒狀劑型。顆粒劑的制備過程通常包括藥材提取純化、濃縮、干燥以及成型等工藝流程。是一種便于攜帶和服用的療效顯著提高的新劑型,具有易溶解、易吸收,生物利用度高、服用方便等特點[11]。
提取工藝是制劑過程中最重要的環(huán)節(jié)之一,對中藥制劑的處方篩選及藥物療效起著根本性作用,傳統(tǒng)提取方法提取效率低,目前中藥復(fù)方提取工藝優(yōu)化主要有正交設(shè)計、均勻設(shè)計及響應(yīng)面設(shè)計等方法,安排多因素、多水平試驗,設(shè)計合理試驗方案[12-13]。本研究先采用單因素變量法考察提取溶媒的種類,再運用正交實驗法以總黃酮含量、槲皮素含量以及出膏率為指標(biāo)進(jìn)行綜合評價,篩選出最佳提取工藝為10倍量65%的乙醇回流提取2次,每次60 min。優(yōu)選工藝穩(wěn)定、可靠。蘇春梅等[15]研究益母草顆粒的制備工藝時,采用正交試驗優(yōu)選出了最佳提取工藝。喬鵬等[16]在研究瓦松顆粒劑制備工藝時,也采用了乙醇連續(xù)回流提取法提取黃酮類有效成分。中藥提取后,要進(jìn)行濃縮成浸膏,是關(guān)系到制粒工藝的關(guān)鍵技術(shù)之一,浸膏相對密度越大,制粒時輔料加入量越少,服用劑量越少。通常采用簡易的真空濃縮。該方法具有高效且有效成分損失小的特點。單因素變量法考察了濃縮溫度對總黃酮含量的影響,確定最佳濃縮溫度為60℃。干燥工藝采用真空減壓干燥工藝,該法具有干燥時間短及干燥溫度低等特點,能有效降低有效成分的。單因素變量法考察了干燥溫度對總黃酮含量的影響,結(jié)果表明,80℃下干燥效率最高。
輔料是制備制劑時必要的物質(zhì),顆粒劑常用的輔料有乳糖、糊精、蔗糖。許玲玲等[17]在研究杉斛顆粒劑制備工藝時,考察了不同的輔料對顆粒劑的影響,發(fā)現(xiàn)淀粉和糊精(1∶1)混合制成的顆粒劑合格率最高。楓蓼顆粒劑的成型工藝輔料篩選中,考慮到成本因素及顆粒的成型性等因素,本研究選用了糊精和蔗糖。采用正交因素試驗進(jìn)行處方篩選出最佳成型工藝為85%乙醇作潤濕劑,藥物與輔料比例為1∶1.5,糊精與蔗糖比例為3∶2制軟材,過14目篩,干燥溫度為60 ℃,14目篩整粒即得。中間體質(zhì)量控制考察了顆粒的溶化性、休止角及臨界相對濕度。休止角測量法為評價顆粒流動性的常用且簡便的方法,此法精確度不高,容易形成誤差,因此要做平行試驗3次,且注入物料是要盡量使其與標(biāo)準(zhǔn)圓錐形狀接近,以減少試驗誤差。王秀麗等[18]在研究慈姑多糖部位顆粒劑制備工藝時,休止角測量3次,平均值為39°,流動性符合要求。本次結(jié)果顯示顆粒全部溶解,休止角平均數(shù)為34.2°,流動性良好,臨界相對濕度為67%。
猜你喜歡
四川蠶業(yè)(2021年3期)2021-02-12 02:38:46
山東冶金(2019年6期)2020-01-06 07:45:54
世界農(nóng)藥(2019年2期)2019-07-13 05:55:12
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
銅業(yè)工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
湖南師范大學(xué)自然科學(xué)學(xué)報(2015年1期)2015-02-27 14:50:04
